Practice Essentials
Wilson disease is a rare autosomal recessive inherited disorder of copper metabolism that is characterized by excessive deposition of copper in the liver, brain, and other tissues (see the image below). Wilson disease is often fatal if not recognized and treated when symptomatic.
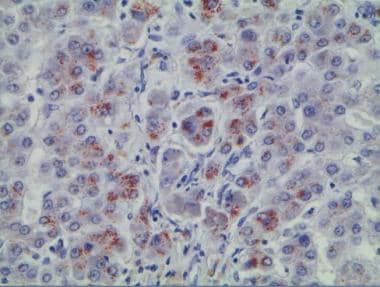 Wilson disease biopsy specimen with rhodanine stain (stain specific for copper deposition).
Wilson disease biopsy specimen with rhodanine stain (stain specific for copper deposition).
Signs and symptoms
Hepatic dysfunction is the presenting feature in more than half of patients. Although the condition may manifest as acute hepatitis, the three major patterns of hepatic involvement are as follows:
-
Chronic active hepatitis
-
Cirrhosis (the most common initial presentation)
-
Fulminant hepatic failure
Signs of fulminant hepatic failure include the following:
-
Ascites and prominent abdominal veins
-
Spider nevi
-
Palmar erythema
-
Digital clubbing
-
Hematemesis
-
Jaundice
Neuropsychiatric features
Most patients who present with neuropsychiatric manifestations have cirrhosis. The most common presenting neurologic feature is asymmetric tremor, which is variable in character and may be predominantly resting, postural, or kinetic.
Frequent early symptoms include the following:
-
Difficulty speaking
-
Excessive salivation
-
Ataxia
-
Masklike facies
-
Clumsiness with the hands
-
Personality changes
Late manifestations (now rare because of earlier diagnosis and treatment) include the following:
-
Dystonia
-
Spasticity
-
Grand mal seizures
-
Rigidity
-
Flexion contractures
Psychiatric features (10%-20% of patients) include the following:
-
Emotional lability
-
Impulsiveness
-
Disinhibition
-
Self-injurious behavior
Psychiatric abnormalities associated with Wilson disease has been divided into the following four basic categories:
-
Behavioral
-
Affective
-
Schizophrenic-like
-
Cognitive
Musculoskeletal manifestations
-
The arthropathy of Wilson disease is a degenerative process that resembles premature osteoarthritis
-
Symptomatic joint disease usually arises late in the course of the disease, frequently after age 20 years
-
The arthropathy generally involves the spine and large appendicular joints (eg, knees, wrists, hips)
-
Osteochondritis dissecans, chondromalacia patellae, and chondrocalcinosis have also been described
Hematologic and renal manifestations
-
Coombs-negative acute intravascular hemolysis (10%-15%)
-
Urolithiasis
-
Hematuria
Kayser-Fleischer rings
-
Formed by the deposition of copper in the Descemet membrane in the limbus of the cornea
-
The color may range from greenish gold to brown
-
Well-developed rings may be readily visible to the naked eye or with an ophthalmoscope set at +40
-
When not visible to the unaided eye, the rings may be identified using slit-lamp examination or gonioscopy
-
Observed in up to 90% of individuals with symptomatic Wilson disease and almost invariably present in those with neurologic manifestations
-
No longer considered pathognomonic of Wilson disease unless accompanied by neurologic manifestations, as they may also be observed in patients with chronic cholestatic disorders
Additional manifestations
-
Skeletal abnormalities (eg, osteoporosis, osteomalacia, rickets, spontaneous fractures, polyarthritis)
-
Cardiac manifestations (eg, rhythm abnormalities, increased autonomic tone)
-
Skin pigmentation and a bluish discoloration at the base of the fingernails (azure lunulae)
See Presentation for more detail.
Diagnosis
Considerations in the workup of Wilson disease are as follows:
-
Serum ceruloplasmin levels are less than 20 mg/dL (reference range, 20-40 mg/dL) in approximately 90% of all patients with Wilson disease
-
The urinary copper excretion rate is greater than 100 mcg/day (reference range, < 40 mcg/day) in most patients with symptomatic Wilson disease, but it may also be elevated in other cholestatic liver diseases
-
In a patient with Kayser-Fleischer rings, a serum ceruloplasmin level < 20 mg/dL and 24-hour urine copper excretion >40 mcg/day establish the diagnosis of Wilson disease
-
Hepatic copper concentration (criterion standard) on a liver biopsy specimen is >250 mcg/g of dry weight even in asymptomatic patients; a normal result (15-55 mcg/g) effectively excludes the diagnosis of untreated Wilson disease, but elevation may be found in other chronic hepatic disorders
-
Radiolabeled copper testing directly assays hepatic copper metabolism
-
Genetic testing is limited to screening of family members for an identified mutation detected in the index patient
-
Brain imaging shows characteristic findings; MRI appears to be more sensitive than CT in detecting early lesions
-
Abdominal imaging findings are neither sensitive nor specific
-
Resting ECG abnormalities include left ventricular or biventricular hypertrophy, early repolarization, ST segment depression, T-wave inversion, and various arrhythmias
-
Electron microscopic detection of copper-containing hepatocytic lysosomes is helpful in the diagnosis of the early stages of Wilson disease, in addition to the quantification of hepatic copper by atomic absorption spectrophotometry
See Workup for more detail.
Management
Features of treatment of Wilson disease are as follows:
-
The mainstay of therapy is lifelong use of chelating agents (eg, penicillamine, trientine)
-
Symptoms, particularly neurologic ones, may worsen with initiation of chelation
-
Surgical decompression or transjugular intrahepatic shunting (TIPS) is reserved for recurrent or uncontrolled variceal bleeding unresponsive to standard conservative measures
-
Orthotopic liver transplantation is curative
Other treatments for Wilson disease include the following:
-
Anticholinergics, baclofen, GABA antagonists, and levodopa to treat parkinsonism and dystonia
-
Antiepileptics to treat seizures
-
Neuroleptics to treat psychiatric symptoms
-
Protein restriction, lactulose, or both to treat hepatic encephalopathy
See Treatment and Medication for more detail.
Background
Wilson disease is a rare autosomal recessive inherited disorder of copper metabolism. The condition is characterized by excessive deposition of copper in the liver, brain, and other tissues. The major physiologic aberration is excessive absorption of copper from the small intestine and decreased excretion of copper by the liver. (See Etiology.) The available evidence suggests that substantial increases in copper concentrations in the central nervous system persist for a long time during chelating treatment and that local accumulation of iron in certain brain nuclei may occur during the course of the disease. [1]
The genetic defect, localized to arm 13q, has been shown to affect the copper-transporting adenosine triphosphatase (ATPase) gene (ATP7B) in the liver. [2] Patients with Wilson disease more often initially present with hepatic manifestations when identified in the first decade of life as compared with more neuropsychiatric illness later, and the latter most commonly occurs during the third decade. The diagnosis is established by no individual test but requires the use of some combination of serum ceruloplasmin level, urinary copper excretion, presence of Kayser-Fleischer rings, and hepatic copper content when biopsy is required. (See Etiology, Presentation, and Workup.)
Although it is extremely rare in clinical practice, Wilson disease is important because it is often fatal if not recognized and treated when symptomatic. Often, the diagnosis is not made until adulthood, despite manifestations of the disease beginning to develop in childhood. (See Differentials, Treatment, and Medication.)
Staging
The natural history of Wilson disease may be considered in four stages, as follows:
-
Stage I - The initial period of accumulation of copper within hepatic binding sites
-
Stage II - The acute redistribution of copper within the liver and its release into the circulation
-
Stage III - The chronic accumulation of copper in the brain and other extrahepatic tissue, with progressive and eventually fatal disease
-
Stage IV - Restoration of copper balance by the use of long-term chelation therapy
Patient education
For patient education information, see the Digestive Disorders Center, as well as Cirrhosis.
Etiology
The normal estimated total body copper content is 50-100 mg, and the average daily intake 2-5 mg, depending on an individual’s intake of legumes, meats, shellfish, and chocolate. Copper is an important component of several metabolic enzymes, including lysyl oxidase, cytochrome c oxidase, superoxide dismutase, and dopamine beta-hydroxylase.
Around 50%-75% of intestinal copper is absorbed and then transported to the hepatocytes. This pathway is intact in Wilson disease. After copper reaches the hepatocyte, it is incorporated into copper-containing enzymes and copper-binding proteins (CBPs), including ceruloplasmin, a serum ferroxidase. Within the liver, the majority of in-infancy (< 6 mo) CBP granules staining positive may be normal. After six months, positive staining of CBPs for copper is almost exclusively found in association with liver diseases such as Wilson disease, chronic biliary disorders (eg, primary biliary cirrhosis, primary sclerosing cholangitis), cirrhosis/extensive fibrosis, and primary liver tumors (most often fibrolamellar hepatocellular carcinoma).
Excess copper may be rendered nontoxic by forming complexes with apo-metallothionein to produce copper-metallothionein, or it may be excreted into bile. Normal copper balance is maintained by regulation of excretion, rather than absorption, and the predominant route of copper excretion (approximately 95%) is hepatobiliary in nature.
In Wilson disease, the processes of incorporation of copper into ceruloplasmin and excretion of excess copper into bile are impaired. [3] The transport of copper by the copper-transporting P-type ATPase is defective in Wilson disease secondary to one of several mutations in the ATP7B gene. [4] By genetic linkage studies, Bowcock and colleagues narrowed the assignment of the Wilson disease locus to 13q14-q21. [5]
Many of the gene defects for ATP7B are small deletions, insertions, or missense mutations. Most patients carry different mutations on each of their 2 chromosomes. More than 40 different mutations have been identified, the most common of which is a change from a histidine to a glutamine (H1069Q). Stapelbroek et al linked the H1069Q mutation to a late and neurologic presentation. [6]
The excess copper resulting from Wilson disease promotes free radical formation that results in oxidation of lipids and proteins. Ultrastructural abnormalities in the earliest stages of hepatocellular injury, involving the endoplasmic reticulum, mitochondria, peroxisomes, and nuclei, have been identified. Initially, the excess copper accumulates in the liver, leading to damage to hepatocytes. Eventually, as liver copper levels increase, it increases in the circulation and is deposited in other organs.
Stuehler et al reported that base pair changes in the MURR1 gene were associated with an earlier presentation of Wilson disease. [7] MURR1 had previously been established to cause canine copper toxicosis in Bedlington terriers.
Epidemiology
In the United States, the carrier frequency is 1 per 90 individuals. The prevalence of Wilson disease is 1 per 30,000 individuals.
Worldwide, the incidence of Wilson disease is 10-30 million cases, and the heterozygote carrier rate is 1 case per 100 persons, with the genetic mutation frequency varying from 0.3%-0.7%. In Japan, the rate is 1 case per 30,000 population, compared with 1 case per 100,000 population in Australia. The increased frequency in certain countries is due to high rates of consanguinity. The fulminant presentation of Wilson disease is more common in females than in males.
Age-related presentations
A German study of patients with Wilson disease illustrated that patients presenting earlier show predominantly hepatic symptoms (15.5 [9.6] y), while those presenting later more often present with neurological symptoms (20.2 [10.8] y). [8]
Thomas and colleagues reviewed the mutations found in the ATP7B gene, and their findings suggested a wide age span in the onset of Wilson disease, perhaps wider than previously considered typical. Mutations that completely disrupt the gene can produce liver disease in early childhood, at a time when Wilson disease may not be considered in the differential diagnosis. [9]
In general, the upper age limit for considering Wilson Disease is 40 years and the lower age limit is 5 years, although the disorder has been detected in children younger than 3 years and in adults older than 70 years. [10]
Prognosis
Patients with a prognostic index (ie, score) of 7 or greater should be considered for liver transplantation (see Table 1, below). All patients in the study associated with this prognostic index who exceeded this score died within 2 months of diagnosis, irrespective of the institution of appropriate medical therapy.
Table. Prognostic Index in Fulminant Wilsonian Hepatitis (Open Table in a new window)
Score |
0 |
1 |
2 |
3 |
4 |
Serum bilirubin (reference range, 3-20 mmol/L) |
< 100 |
100-150 |
151-200 |
201-300 |
>300 |
Serum aspartate transaminase (reference range, 7-40 IU/L) |
< 100 |
100-150 |
151-200 |
201-300 |
>300 |
Prothrombin time prolongation (seconds) |
< 4 |
4-8 |
9-12 |
13-20 |
>30 |
Prognosis after liver transplantation is relatively good. In a study involving 55 patients with Wilson disease who underwent hepatic transplantation, the 1-year survival rate was 79% and the overall survival rate was 72% at 3 months to 20 years. Another study of 32 patients reported a 1-year survival of 90.6%, a 5-year survival rate of 83.7%, and a 10-year survival rate of 79.9% after living donor liver transplantation. [11]
Important clues for the diagnosis of Wilson disease that a clinician must recognize are a younger patient with hemolytic anemia, impaired hepatic synthetic function, and normal alkaline phosphatase values.
Complications
The major complications in patients with untreated Wilson disease are those associated with acute liver failure, chronic hepatic dysfunction with either portal hypertension or hepatocellular carcinoma, and the sometimes-relentless course to cirrhosis, which is characterized by a progressive lassitude, fatigue, anorexia, jaundice, spider angiomas, splenomegaly, and ascites. Bleeding from varices, hepatic encephalopathy, hepatorenal syndrome, and coagulation abnormalities occur as liver failure ensues. Death occurs, generally at age 30 years, if emergent liver transplantation is not performed.
Unfortunately, Wilson disease has other systemic consequences of copper overload. Most patients who present with neuropsychiatric manifestations have cirrhosis. The reported percentage of patients with psychiatric symptoms as the presenting clinical feature is 10%-20%. The range of psychiatric abnormalities associated with Wilson disease extends from behavioral/mood state disturbances through movement disorders (occasionally choreoathetoid) or parkinsonian features. These features, on occasion, can be made worse with chelation therapy.
-
Computed tomography (CT) scan in a 15-year-old boy who presented with central nervous system findings consistent with Wilson disease. The CT scan reveals hypodense regions in the basal ganglia (caudate nucleus, putamen, globus pallidus). The differential diagnosis based on this image alone included leukodystrophy, vasculitis, and, less likely, infection. Ventricular enlargement and posterior fossa atrophy may also be seen on brain CT scans in a patient with Wilson disease. The extent of involvement as depicted on CT scans does not provide prognostic information.
-
Approach to the diagnosis of Wilson disease (WD) in a patient with unexplained liver disease. KF = Kayser-Fleischer ring; CPN = ceruloplasmin. From the American Association for the Study of Liver Diseases Practice Guidelines.
-
In this particular case, there is abundant Mallory hyaline. Another notable finding is the moderate to marked chronic inflammation which involved most portal tracts and periportal/perinodular areas.
-
Prismaflex eXeed II adds citrate anticoagulation with integrated calcium management. Image courtesy of Gambro.
-
Molecular adsorbents recirculating system (MARS) circuit.
-
Biopsy specimen showing hepatocellular injury in an explant specimen from a patient transplanted for Wilson Disease.
-
Biopsy specimen showing a more detailed image of the cellular injury in acute Wilson disease.
-
Wilson disease biopsy specimen with rhodanine stain.
-
Wilson disease biopsy specimen with rhodanine stain (stain specific for copper deposition).
Tables
Score |
0 |
1 |
2 |
3 |
4 |
Serum bilirubin (reference range, 3-20 mmol/L) |
< 100 |
100-150 |
151-200 |
201-300 |
>300 |
Serum aspartate transaminase (reference range, 7-40 IU/L) |
< 100 |
100-150 |
151-200 |
201-300 |
>300 |
Prothrombin time prolongation (seconds) |
< 4 |
4-8 |
9-12 |
13-20 |
>30 |