Background
Ataxia is defined as “an inability to coordinate voluntary muscular movements.” Ataxia describes a neurologic symptom that can be seen in a myriad of diseases and conditions. Ataxia can be progressive or static, and can present at any age. Identifying the etiology of ataxia can be a complex task. For example, ataxia may be caused by a lack of proprioception and processing of environmental information by the extremities, which makes ambulation difficult as the feet do not know where they are in space. This phenomenon is called a sensory ataxia, as can be seen in patients with peripheral neuropathies. Alternatively, vestibular dysfunction can also cause ataxia, such as patients with Benign Paroxysmal Positional Vertigo (BPPV). Ataxia may be caused by dysfunction of the cerebellum, the part of the brain that coordinates movements of muscles and maintains the body’s equilibrium. Common etiologies of cerebellar dysfunction include acquired forms (which can be related to nutritional, immunologic, or degenerative causes) and inherited causes (related to genetics). Differentiating acquired from inherited forms of ataxia can help determine the expected disease course, the etiology of the condition, treatment options that may be available, and assist in genetic counseling. Inherited etiologies involve a genetic or biochemical defect which leads to the formation of ataxia. A positive family history of similar conditions, physical exam findings, neuroimaging, and genetic testing can help make the diagnosis of an inherited ataxia.
This article reviews the current understanding of inherited neurologic and metabolic disorders manifesting with ataxia as a clinical feature. It will highlight key clinical features, diagnosis, and pathophysiologic insights gleaned from molecular genetic studies, as well as current treatment strategies.
In general, hereditary genetic and metabolic disorders involve the nervous system at multiple levels, resulting in varied manifestations. Common clinical presentations of such disorders in childhood include the following features in combination:
-
Developmental delay
-
Neurologic or developmental regression
-
Family history of similar symptoms in a sibling or closely related individual
-
Episodic alteration in level of consciousness or recurrent neurologic symptoms
-
Multisystem involvement (in addition to neurologic systems)
-
Development and progression of a particular neurologic sign such as ataxia or seizures
Neurologic symptoms and signs such as seizures and movement disorders (eg, dystonia, chorea) may accompany the disorders that cause inherited ataxias. A wide range of molecular defects have been identified in which the spinocerebellar pathways are involved. [1, 79] Consequently, many variations are encountered in the clinical phenotype, ranging from findings of pure cerebellar dysfunction to mixed patterns of involvement reflecting extrapyramidal pathways, brainstem, and/or cerebral cortical involvement. [78]
Despite this remarkable diversity of genetic defects and mechanisms, the pathologic responses within the nervous system are limited in terms of the targeted pathways. This feature likely contributes to the significant overlap seen in the clinical presentation. Nevertheless, delineation of the clinical phenotype represents an important first step in the diagnostic process. The clinical phenotype guides the geneticist in a search for appropriate diagnostic tests, reducing costs of laboratory workup.
The group of disorders manifesting with ataxia is expanding constantly as the genetic basis for many of the inherited ataxias are unraveled (for example, there are around 45 spinocerebellar ataxias [SCAs] currently recognized). Study of subcellular organelle structures has enabled delineation of aspects of mitochondrial, lysosomal, and peroxisomal disorders. However, despite the advances in the understanding of pathogenesis, there has been a lag in the development of effective treatments for this group of disorders. [2]
As the underlying mechanisms of disease begin to be understood, the inherent challenges are apparent; for instance, several ataxias are caused by defects in DNA repair, while others may result from protein folding and chaperoning defects. Advances in genomics, proteomics, transcriptomics, and metabolomics are paving the way towards understanding of gene function, protein synthesis and transcription, and gene-gene and protein-protein interactions. These studies hopefully will provide the basis for a new set of designer drugs geared towards individualized treatments.
The cerebellum and its pathways in health and disease
The main functions of the cerebellum are related to locomotion, postural control, voluntary movements, and cognition within the cerebellum. The subsequent few sections will focus upon these main functions, the anatomical pathways, and the underlying structures. Understanding their functions will aid in highlighting the clinical manifestations of lesions in these structures.
Locomotive functions of cerebellum
The cerebellum has a crucial role in balance and locomotion. Functional specificity allows regions of the cerebellum to control aspects of motor control. These anatomical-functional relationships are discussed below.
-
The medial zone of cerebellum: This zone integrates spinal and vestibular inputs and subsequently projects out through the fastigial nucleus to vestibulospinal and reticulospinal tracts. These regions appear to exert modulatory control of the rhythmic flexor and extensor locomotor pattern generated by vestibular and reticular nuclei. These connections also control extensor tone to maintain upright balance and stance. A lesion in this zone leads to a significant balance problem and impairment of postural tone.
-
Intermediate zone (paravermal region): This zone receives input from the spine (via spinocerebellar tracts) and projects out through the globose and emboliform nuclei to the red nucleus and cerebral cortex. It integrates spinal and cortical inputs and influences locomotion through projections to motor cortical areas. The main function of this region is related to specific control of limb placement including timing, elevation and trajectory of limb elevation, and descent. Damage to this region leads to gait ataxia and swing phase overshoot of legs but no overt change in balance or postural tone.
-
Lateral zone: This area receives input primarily from cerebral cortical area via pontine nucleus (corticopontocerebellar fibers) and projects out via the dentate nucleus through the red nucleus to the thalamus and cerebral cortical areas. This zone influences motor activities via cortical interactions and has an important role in voluntary modification of motor activities and the locomotor cycle. Lateral cerebellum is especially active in novel walking conditions where precise limb placement is necessary. It modulates visually guided motor activities because of the robust projection it receives from the visual cortex. A lesion in this region leads to limb ataxia and locomotion problems in novel and challenging situations. During uninterrupted walking, balance deficits contribute much more strongly to cerebellar gait ataxia (medial and intermediate zone) than do visually guided leg control deficits seen in the lateral zone.
Postural sway with a cerebellar lesion
Cerebellar damage in humans typically results in postural sway. Balance deficits as a result of lesions in midline cerebellar structures (vestibulocerebellum) lead to low frequency, high amplitude postural sway without a preferred direction and without intersegmental movements. On the other hand, in those with lesions in the intermediate zone (including anterior lobe), balance deficit is characterized by increased postural sway of high velocity and low amplitude; anteroposterior direction; postural tremor; and increased intersegmental movements of the head, trunk, and legs. Subjects with lesions in the lateral zone have only slight postural instability or sway.
Cerebellar control of voluntary movements
Cerebral cortical association areas modulate voluntary movements that are executed by the motor cortex. Motor cortex may act as a controller driving lower motor neurons in the brain stem and spinal cord. But there is a robust cerebellocerebral loop that modulates these motor functions as well. These loops connect the intermediate part of the cerebellum to the association cortex and the motor cortex. In turn, the outputs from the intermediate zone of the cerebellum converge down to meet the cerebral output at red nucleus and olive. Thus, both loop and parallel pathways exist between the cerebrum and cerebellum. The cerebellum influences voluntary activities through these pathways.
One of the major functions of the cerebellum is motor adaptation based on trial and error practice (error driven learning mechanism). The process takes place through long-term depression (LTD), a characteristic form of synaptic plasticity occurring at parallel fiber-Purkinje cell synapses. [6, 7]
Cognitive function of cerebellum
A closed cerebellocerebral loop is found in the prefrontal cortex and thus the cerebellum provides a forward model for mental functions in the cerebral cortex. This is analogous to already discussed cerebellocerebral loop concerned with motor functions. A primary cerebellar injury in premature infants has shown to be associated with contralateral decrease in cerebral volume. [8] This strengthens the importance of the cerebellocerebral connections responsible for important cognitive functions.
A mental concept of an image or idea is formed in the temporoparietal association cortex. These already formed mental concepts are manipulated by the prefrontal cortex. After repeated exercise, the cerebellum copies a mental model to form an internal model through cerebello-cerebral loop. Because of this internal model formed by the cerebellum, we are able to conduct movements and thoughts unconsciously (processes occurring in the cerebellum are felt to not reach awareness). For this reason, when an idea "just comes out of the blue"it is possible that it is related to this pathway. However, the cognitive contributions of the cerebellum is still debated. [114, 115]
Localization overview
As demonstrated above, the localization and regional distribution of pathology within the cerebellum dictates the clinical findings. Lesions of the midline cerebellar vermis produce truncal and gait ataxia, while involvement of the lateral cerebellar hemispheres produces a limb ataxia. Interruption of afferent and efferent connections within the neocerebellar system results in an ataxic gait (ie, swaying in the standing posture, staggering while walking with a tendency to fall, and the adoption of a compensatory wide base), scanning dysarthria, explosive speech, hypotonia, intention tremor (ie, oscillation of limbs that is pronounced at the end of a planned movement), dysdiadochokinesia (ie, impaired alternating movements), dysmetria (ie, impaired judgment of distance), decomposition of movement, and abnormalities of eye movements (ie, nystagmus).
Clinical phenotypes show considerable overlap; however, the genetic, molecular, and biochemical causes for these disorders are often distinct. Some phenotypes (dominant ataxias) show considerable genetic heterogeneity. These phenotypes may manifest with pure ataxia or involve multiple levels of the nervous system (including dementia, seizures, disturbance in proprioceptive function, movement disorders, and polymyoclonus).
Genetic-biochemical basis for classification
Early attempts to classify inherited ataxias were based on anatomic localization of pathologic changes (eg, spinocerebellar, pure cerebellar). In 1993, Harding introduced another classification in which the ataxias were placed into 3 categories, congenital, inherited metabolic syndromes with known biochemical defects, and degenerative ataxias of unknown cause. [9] The last category was subdivided further into early onset (< 25 y) and late-onset types. Although widely accepted, this classification does not incorporate or reflect current understanding of this group of disorders.
Although ataxia is a prominent feature of all these disorders, the presentation can be variable (eg, static vs progressive, intermittent vs chronic, early vs delayed). The mode of inheritance also varies. Autosomal dominant, recessive, and nonmendelian inheritance patterns have been described. Nonmendelian inheritance patterns have become increasingly significant in the understanding of the biology of human diseases. The term refers to disorders of inheritance for which the rules of Mendelian genetics do not apply. Disorders of triplet repeat expansion and certain mitochondrial defects are examples of nonmendelian inheritance.
Clearly, a revision of the classification of hereditary ataxias is necessary to include current concepts. Such a classification system is obviously an evolving one, with a separate category that includes those disorders where the molecular basis is presently unknown. Selected conditions in each category are discussed below. The following outline includes clinical features and known information about gene products and known or putative function. Treatment options are only included where specific measures are available. The reader interested in the specifics of different conditions is referred to one of several excellent reviews on the subject in the Reference section.
Classification using a genetic-biochemical basis is as follows:
-
Nonprogressive ataxias [10]
Pure congenital cerebellar ataxias with or without cerebellar hypoplasia
Autosomal recessive
Autosomal dominant
X-linked
Unknown
With posterior fossa malformations - Autosomal recessive (eg, Dandy Walker syndrome)
Congenital ataxia syndromes with cerebellar malformations
Autosomal recessive (eg, Joubert syndrome)
X-linked recessive (eg, X-linked congenital cerebellar hypoplasia and external ophthalmoplegia)
-
Intermittent/episodic ataxias
Autosomal dominant - Channelopathies (eg, episodic ataxias [EA] 1, EA 2])
Autosomal recessive - Enzyme defects (eg, maple syrup urine disease [MSUD], urea cycle defects)
X-linked - Enzyme defects (eg, ornithine transcarbamylase [OTC] deficiency)
-
Progressive ataxias with or without multisystem involvement
Autosomal dominant - Ataxias with spinocerebellar dysfunction
Triplet repeat disorders and polyglutamine accumulation (eg, SCAs 1-23, dentatorubropallidoluysian atrophy [DRPLA])
Autosomal recessive
Triplet repeat disorders (eg, Friedreich ataxia)
Impaired DNA repair mechanisms (eg, xeroderma pigmentosum, Cockayne syndrome)
Enzyme defects (eg, Refsum disease, sphingolipidosis)
Protein misfolding (eg, spastic ataxia of Charlevoix-Saguenay)
Maternal inheritance - Mitochondrial disorders (eg, neuropathy, ataxia, retinitis pigmentosa [NARP])
-
Ataxias with polymyoclonus and seizures
Autosomal recessive
Dodecamer repeat expansions (eg, Baltic myoclonus)
Enzyme defects (eg, neuronal ceroid lipofuscinosis)
Maternal inheritance - Mitochondrial cytopathies (eg, myoclonic epilepsy with ragged-red fiber disease [MERRF])
-
Other (unidentified mechanisms)
Angelman syndrome
Fragile X–related ataxia/tremor
In summary, the authors suggest a system of classification based on clinical features as the first distinction, mode of inheritance as the second distinction, and pathogenetic mechanisms as the third distinction. Although far from an ideal system, it serves to bring some order into a heterogeneous group of disorders. Clearly the classification is an evolving process because some disorders could be considered in more than one tier, eg, mitochondrial cytopathies can manifest with myoclonic epilepsy and ataxia, as well as chronic progressive ataxia as in the NARP syndrome.
Molecular Genetics and Putative Mechanisms of Cerebellar Disease
The mechanisms underlying disorders with cerebellar ataxia as a symptom reflect the diversity of etiologies that have been identified. For instance, genetic mutations affecting ion channel structure and function cause both intermittent and chronic symptoms, [11] and recessively inherited enzymopathies (enzyme deficiency) cause symptoms through accumulation of neurotoxic storage material and/or precursor metabolites. The understanding of mechanisms of neurodegeneration resulting in cerebellar disease has been influenced by discoveries in the molecular genetics of nontraditional inheritance patterns underlying conditions such as SCAs and mitochondrial disorders. Therefore, special aspects of molecular genetics and putative mechanisms of cerebellar disease are discussed together (see image below).
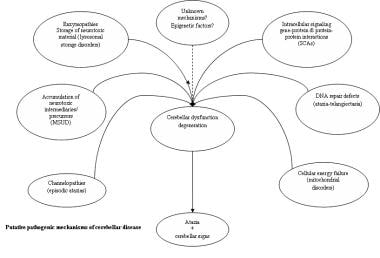
Triplet repeat expansions
This class of mutation is characterized by dynamic expansion of tandem nucleotide repeats in the human genome. These stretches of repeats tend to be inherently unstable, and this instability favors expansion. When the length of the repeat expansion exceeds the range in the general population, a symptomatic state may result. These mutations help explain clinical observations of increasing severity of symptoms and an earlier age of onset in successive generations seen with several of the dominantly inherited disorders—a phenomenon termed genetic anticipation. Such dynamic mutations form the basis of an increasing list of inherited neurologic disorders that includes intellectual disability (fragile X syndrome), myotonic dystrophy, oculopharyngeal muscular dystrophy, Friedreich ataxia, Huntington disease, and the dominantly inherited cerebellar ataxias.
The trinucleotide expansion of cytosine, adenine, and guanine (CAG) repeats is translated into a polyglutamine tail, a common feature of several of the dominantly inherited ataxias. The expansion above a critical threshold, which appears to be different for each SCA type, determines presence of disease. The causative proteins for each type bear no homology to other known proteins or to each other apart from the polyglutamine tail. The polyglutamine tails themselves appear to be toxic once a disease-specific threshold is reached, and this central feature suggests a final common pathway.
The pathogenic mechanism(s) underlying cerebellar disease appear to involve proteolytic cleavage and nuclear accumulation of toxic products. Such proteolytic cleavage, by releasing toxic fragments containing an expanded polyglutamine tail, may serve to further facilitate entry of cytoplasmic polyglutamine proteins to the nucleus. Secondary processes for neuronal injury likely involve downstream effects of apoptotic activation, accumulation, misfolding, aggregation, and sequestration of other proteins such as transcription factors and chaperones, leading to dysfunction of proteins and their intranuclear or intracellular accumulation. The putative disease mechanisms involved in the SCAs can be categorized into the following:
-
Transcriptional abnormalities (SCA17 and SCA7): Ataxins appear to function as transcriptional regulators, and the interaction with polyglutamine proteins results in an impairment of transcription. At other times, transcription factors may be sequestered into the polyglutamine aggregates, leading to transcriptional shutdown and neuronal death.
-
Calcium signaling defects (SCA6 and SCA14): In SCA6, the expanded CAG repeat is within a gene coding for the alpha subunit of the voltage-gated calcium channel. The polyglutamine aggregates in this disorder are cytoplasmic, and altered channel function may be responsible rather than a toxic gain in function.
-
Phosphorylation defects (SCA12 and SCA14): In these disorders, protein phosphorylation mediated through specific enzymes belonging to serine/threonine phosphatase (SCA12) and serine threonine kinase (SCA14) families are affected. A wide variety of cellular signaling pathways where these function as second messengers can be secondarily affected.
-
Defective ubiquitination and proteosome function (SCA3): Protein handling and clearance in the cell is affected through the ubiquitin-proteosome pathway. Components of this pathway may get sequestered in the polyglutamine aggregates, leading to a perturbation in cellular protein homeostasis.
-
Protein misfolding and chaperone defects: Protein folding and structure are vital to normal function; chaperone proteins facilitate this folding properly. Dysfunction of chaperone proteins may contribute to protein misfolding. Such a process may underscore the pathogenic mechanism in SCA1, in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), and in the leukoencephalopathy associated with vanishing white matter (VWM).
Mitochondrial DNA defects
Since mitochondria were established to carry unique functions through their own functional genome, a new mechanism of nonmendelian inheritance, maternal inheritance, was discovered. All the mitochondria in the newly formed zygote are derived from the ovum (ie, maternally derived). Mitochondrial disorders can result from defects of mitochondrial proteins, either coded by the nuclear or by the mitochondrial DNA (mt DNA). Mitochondrial DNA is more vulnerable to mutations in the oxidizing environment of mitochondria because its repair mechanisms are poor compared to nuclear DNA. Mutations in mitochondria accumulate in cells until a threshold is reached. Eventually, the proportion of mutant mitochondria exceeds wild type, resulting in the manifestation of impaired cell function. [12]
The process of uneven replicative segregation ensures different proportions of mutant and wild types in different tissues, a condition termed heteroplasmy. Mildly-to-moderately deleterious mutations can persist and be transferred to offspring. The differential segregation and production of reactive oxygen species can vary among tissues and organ systems in affected individuals, giving rise to varying phenotypes.
Postmitotic cells such as neurons appear to carry higher ratios of mutant mitochondrial DNA, which thereby confer vulnerability to metabolic stress. This vulnerability may show a regional variation within the different regions of the brain, thereby partially explaining the variable patterns of neurologic involvement in many mitochondrial disorders. Some of the examples of mitochondrial disorders manifesting with ataxia include Friedreich ataxia (GAA repeat expansion-nuclear), MELAS syndrome ([mitochondrial myopathy, encephalopathy, lactacidosis, stroke syndrome] A3243-G mutation-maternal), ataxia with selective vitamin E deficiency (AVED), and X-linked ataxia with sideroblastic anemia. [13]
DNA repair defects
Mutations in proteins involved in repairing DNA breaks seem to provide yet another pathway resulting in disorders with ataxia (eg, ataxia -telangiectasia, ataxia with oculomotor apraxia types 1 and 2, SCA with sensory neuropathy [SCAN1]). The ataxia telangiectasia mutated (ATM) protein functionally belongs to a family of protein kinases with the critical role of rapidly healing DNA breaks. Mutations in this protein cause ataxia telangiectasia. Aprataxin, a histidine triad protein is involved similarly in single-stranded DNA repair, while senataxin is involved in splicing and termination of tRNA and may also function as a DNA helicase. [14]
Nonprogressive Cerebellar Ataxias
This group includes diverse conditions that manifest either at birth or in early life. A structural abnormality in the form of cerebellar hypoplasia with or without other posterior fossa malformations affecting the brainstem structures may or may not be demonstrable. Because of the complex maturational and myelination processes within the brain that are age related, the clinical presentation of these disorders in early life is marked by symptoms other than ataxia. Most often hypotonia and developmental delays are striking. Ataxia is only recognized when efforts at independent walking are unsuccessful. In early life, considerable overlap of the neurologic phenotype occurs.
The classification of nonprogressive ataxias is challenging. At the risk of oversimplification, the hereditary nonprogressive ataxias may be categorized as the following:
-
Pure congenital cerebellar ataxias
-
Cerebellar ataxias associated with posterior fossa malformations
-
Congenital ataxic syndromes
-
Ataxic syndromes without cerebellar malformations
The principal differential diagnosis needs to include metabolic and neurodegenerative conditions manifesting in early life, which is further discussed in this article. The suggested metabolic testing and neuroimaging studies can help distinguish this category from other hereditary conditions that are progressive in nature.
A long list of conditions is reported featuring ataxia in association with other clinical features. A few conditions such as Gillespie syndrome include 1 or 2 additional features (eg, intellectual disability, partial aniridia), while other conditions such as Joubert syndrome (ie, hypotonia, hyperventilation, facial dysmorphism, retinal dystrophy, renal involvement) and COACH syndrome (ie, cerebellar hypoplasia, oligophrenia, ataxia, coloboma, hepatic fibrosis) feature malformations in multiple organ systems. Inheritance patterns are usually autosomal recessive or X linked depending on the syndrome. In the case of Joubert syndrome, evidence for genetic heterogeneity exists. Currently, there are at least 34 subtypes of Joubert syndrome due to different causative genes or clinical features. [80]
Table 1. Nonprogressive Congenital Ataxias (Open Table in a new window)
Disorder/Syndrome |
Phenotype* |
Inheritance |
NPCA with or without cerebellar hypoplasia |
Early hypotonia Delayed motor and speech development |
Autosomal recessive
Autosomal dominant
X-linked recessive
Sporadic |
NPCA with posterior fossa malformations (eg, Dandy Walker syndrome) |
Variable association with hydrocephalus Delays in motor development Cognitive delay |
N/A |
Ataxia syndromes, multiple congenital anomalies, and cerebellar hypoplasia (eg, Joubert syndrome, Varadi syndrome, COACH syndrome) |
Encephalo-oculo-hepato-renal anomalies with recognized association patterns of anomalies |
Autosomal recessive
Autosomal dominant
X-linked |
Ataxia syndromes with cerebellar hypoplasia (eg, Gillespie syndrome) |
Partial aniridia Hypogonadotrophic hypogonadism External exophthalmoplegia |
Autosomal recessive |
*Gait ataxia is a constant feature. |
||
Clinical features
See the list below:
-
Early hypotonia
-
Developmental delay
-
Feeding difficulties and oromotor dysfunction
-
Speech delay secondary to articulatory difficulties
-
Cognitive difficulties (may be recognized at a later age)
-
Specific pattern of inheritance upon genetic assessment of the family
Diagnosis
See the list below:
-
Genetic mutation tests: These are available only in selected conditions, eg, certain forms of Joubert syndrome. Testing is available for at least 4 of the causative genes in which mutations appear in Joubert syndrome: AHI1, CEP290, NPHP1, TMEM67, OFD1, C5orf42.
-
Metabolic screening: Results are negative.
-
Neuroimaging studies: MRI is superior because it permits better visualization of the posterior fossa. Variable degrees of hypoplasia of the cerebellar vermis are reported. In more severe cases, the entire vermis may be absent, and associated abnormalities are noted in the cerebellar hemispheres. However, in mild cases, the cerebellum is morphologically normal on imaging studies. Associated abnormalities of the brainstem and supratentorial structures may be of additional value in the diagnosis of syndromes such as Dandy Walker malformation. In Joubert syndrome, a characteristic neuroimaging finding of the "molar-tooth" sign is helpful.
Intermittent or Episodic Ataxias
Channelopathies
Channelopathies represent a number of neurologic disorders that manifest with symptoms of an episodic or transient nature. [15] The underlying molecular defect affects the functioning of a voltage-gated ion channel, thereby altering membrane excitability in neurons. External stimuli often trigger symptoms or episodes. Clinical and genetic heterogeneity is evident in the episodic ataxias with up to 6 forms currently recognized. So far the mutations appear to involve ion channel subunits.
Episodic ataxia 1
See the list below:
-
Gene, inheritance, and pathogenesis: EA1 is a rare autosomal dominant disorder and represents a channelopathy. It is caused by point missense mutations that affect the human voltage-gated potassium channel (KCNA1 gene on band 12p13). This channel is widely expressed, but is especially prominent in the cerebellum. The mutation can impair channel function by reducing the amplitude of the potassium current and by altering its voltage-dependent kinetics. [16, 17]
-
Clinical features
Continuous myokymia between attacks
Duration of seconds to minutes
Partial epilepsy (some individuals in affected families)
Sudden episodes of ataxia precipitated by movement, startle, or emotion
-
Diagnosis
Electroencephalography (EEG) may show continuous rhythmic muscle discharge artifact, which may become more prominent with hyperventilation.
Electromyography is the only helpful investigation; it usually demonstrates continuous motor unit activity in all patients.
Genetic testing can identify the heterozygous patogenic variant in KCNA1.
-
Treatment: Partial responses to acetazolamide, carbamazepine, phenytoin, and phenobarbital have been reported.
Episodic ataxia 2
See the list below:
-
Gene, inheritance, and pathogenesis: EA2 is an autosomal dominant disorder that has been associated with mutations that affect the calcium channel (CACNA1A [18] ) gene at the 19p13 locus. It is allelic to familial hemiplegic migraine and SCA6, wherein mutations affecting the same gene have been described. Haploinsufficiency may underlie the EA2 pathogenesis because the majority of the mutations causing EA2 result in nonfunctional calcium channels. EA2 exhibits incomplete penetrance and variable expressivity both between and within families. [16]
-
Clinical features
Headache (in some families)
Intermittent episodes of ataxia
Absence of myokymia
Provoking factors - Stress, exercise, and fatigue, among others
-
Diagnosis: CACNA1A gene mutation testing is available in certain laboratories
-
Treatment: A few patients with EA2 may respond to acetazolamide
Episodic ataxia 3
A clinically distinct form of autosomal dominant episodic ataxia occurs in the Canadian Mennonite population. The candidate gene maps to a 4 cM region on chromosome 1q42 between markers D1S2712 and D1S2678. No mutations have been identified to date.
-
Clinical features
Adult onset
Vestibular ataxia, vertigo, tinnitus, and interictal myokymia
Symptoms triggered by sudden movement, stress, exertion, and fatigue
-
Diagnosis: No gene tests are available
-
Treatment: The condition responds well to acetazolamide
Episodic ataxia 4
See the list below:
-
Gene, inheritance: This is a very rare autosomal dominant condition. Unknown gene and pathogenesis.
-
Clinical features:
Intermittent episodes of ataxia, vertigo, diplopia that last hours typically
Symptoms provoked by sudden movement, stress, exertion, fatigue
Adult onset
-
Diagnosis: No gene tests are available.
-
Treatment: Not responsive to acetazolamide
Episodic ataxia 5
See the list below:
-
Gene, inheritance, and pathogenesis: Autosomal dominant. Mutated gene is CACNB4 at locus 2q23.3. Unknown pathogenesis.
-
Clinical features
Intermittent vertigo and ataxia lasting several hours
Interictal examination with spontaneous downbeat and gaze-evoked nystagmus, mild dysarthria, and truncal ataxia
-
Diagnosis: CACNB4 mutation
-
Treatment: Favorable response with acetazolamide
Episodic ataxia 6
See the list below:
-
Gene, inheritance, and pathogenesis: EA6 is an autosomal dominant disorder that has been associated with missense mutations in the SLC1A3 gene. It affects the excitatory amino acid transporter 1 (EAAT1), a glial glutamate transporter.
-
Clinical features
Recurrent episodes of ataxia, seizures, migraine and alternating hemiplegia
Attacks provoked by emotional stress, fatigue and alcohol or caffeine intake
-
Diagnosis: Mutation in the SLC1A3 gene
-
Treatment: Responds well to acetazolamide
Episodic ataxia 7
See the list below:
-
Gene, inheritance, and pathogenesis: Inheritance is autosomal dominant. Gene locus is 19q13, but gene is currently unknown.
-
Clinical features
Episodes of ataxia, vertigo, dysarthria and weakness
Onset before the age of 20
Symptoms lasted hours to days and were provoked by exercise and excitement
-
Diagnosis: Unknown
-
Treatment: Unknown
Table 2. Episodic Ataxias (Open Table in a new window)
Disorder/Syndrome |
Phenotype* |
Inheritance |
Locus/Gene |
EA1 |
Intermittent ataxia |
Autosomal dominant |
12q13 KCNA1 |
EA2 |
Intermittent ataxia |
Autosomal dominant |
19q13 CACNA1A |
EA3 |
Intermittent ataxia with vertigo and tinnitus |
Autosomal dominant |
1q42 |
| EA4 | Intermittent ataxia, vertigo, diplopia | Autosomal dominant | Unknown |
| EA5 | Intermittent vertigo and ataxia lasting several hours | Autosomal dominant | 2q23.3 CACNB4 |
| EA6 | Intermittent ataxia, seizures, migraine and alternating hemiplegia |
Autosomal dominant | 5p13.2 SLC1A3 |
| EA7 | Vertigo, weakness, dysarthria | Autosomal dominant | 19q13 |
Inherited Enzyme Defects
Inherited enzyme defects are discussed below.
Maple syrup urine disease (intermittent form)
A delayed presentation of this autosomal recessive form of a branched chain aminoacidopathy may occur at any age from infancy to adulthood. [19, 20, 21, 22, 23]
-
Gene, inheritance, and pathogenesis: This is an autosomal recessive disorder caused by a deficiency of branched chain alpha keto acid dehydrogenase complex. Mutations of at least 4 gene loci are known to result in this condition, including BCKDHA on chromosome 19q13.2, BCKDHB on chromosome 6q14.1, and DBT on chromosome 1p21.2.
-
Clinical features
Characteristic urine odor of maple syrup, as well as in other body fluids and earwax
Intermittent bouts of ataxia and neurologic obtundation progressing to coma
Possibly, intellectual disability and motor delay in intermediate form
-
Diagnosis
Elevation of branched chain amino acids and branched chain keto acids in the urine, plasma, and cerebrospinal fluid (CSF)
Metabolic acidosis, ketonemia, and ketonuria; occasional hypoglycemia and hypoalaninemia
L-alloisoleucine in body fluids (pathognomonic) [24]
Assay of branched chain keto acid dehydrogenase activity in skin fibroblasts
Mutation testing
-
Treatment [25]
Treatment includes restriction of dietary protein intake and supplementation of branched chain amino acid–free synthetic formula to meet protein and other dietary needs.
Begin thiamine supplementation in thiamine-responsive individuals (5–20 mg/kg/d, not to exceed 100 mg/d) immediately. In adults, 100 mg may be administered immediately in the acute situation, followed by further supplementation of 50-100 mg/d until adequate oral intake and a stable clinical state are achieved.
Hartnup disease
The incidence based on neonatal screening data is estimated at 1 in 30,000. The reduced availability of tryptophan may lead to a secondary deficiency of the vitamin niacin (nicotinic acid). [26, 27]
-
Gene, inheritance, and pathogenesis: The locus associated with Hartnup disease is 5p15. This autosomal recessive disorder is caused by defective intestinal transport and renal tubular reabsorption of neutral amino acids (primarily tryptophan). Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. SLC6A19 is a sodium-dependent and chloride-independent neutral amino acid transporter, expressed predominately in kidney and intestine.
-
Clinical features
Intermittent ataxia and other cerebellar signs
Neuropsychiatric dysfunction ranging from emotional lability to frank psychosis
Pellagralike skin rash induced by exposure to sunlight
-
Diagnosis
Excessive excretion of monoamino-monocarboxylic amino acids in urine
Urinary indoxyl derivatives (5-hydroxyindoleacetic acid) detectable in urine following an oral tryptophan load
-
Treatment: Treatment includes a high-protein diet. Niacin supplementation reverses the skin and neuropsychiatric manifestations. A tendency exists for spontaneous improvement.
Pyruvate dehydrogenase deficiency
See the list below:
-
Gene, inheritance, and pathogenesis: The commonest form of pyruvate dehydrogenase (PDH) deficiency is an X-linked recessive disorder that affects a mitochondrial multienzyme complex, which is involved in the conversion of pyruvate to acetyl-CoA. The PDHA1 gene codes for 3 enzymes of the PDH complex. The E1 alpha1 subunit of this complex is most often affected. Inheritance is X linked for the latter form. A high proportion of heterozygous females manifest severe symptoms (in the X-linked form).
-
Clinical features
Many present in early infancy with a catastrophic neurologic picture of hypotonia, lactic acidosis, and seizures (associated with cerebral malformations).
About 30% present with facial dysmorphic features, including microcephaly, narrow head, frontal bossing, long philtrum, episodic ptosis, abnormal eye movements, wide nasal bridge, upturned nose, and flared nostrils.
A benign late-infantile variant can occur.
Episodic ataxia is characteristic.
Uncommonly, mental and motor development is normal.
Fatigue is noticed after exercise.
Transient paraparesis is a feature.
-
Diagnosis
Serum and CSF lactic acidosis is characteristic. The lactate-to-pyruvate ratio is normal.
PDH activity in skin fibroblasts is reduced.
Mutation testing is available in certain laboratories only.
In the prenatal and early infantile form, multiple areas of necrosis in the gray matter, white matter, and basal ganglia are noted on imaging studies.
-
Treatment
Thiamine supplementation in high doses (5–20 mg/kg/d, not to exceed 100 mg/d in acute stage) may be effective in the thiamine-responsive form of the disease.
A ketogenic diet has been effective in some patients.
- Dichloroacetate may help resolve lactic acidosis, however it does not improve neurological damage. Additionally, peripheral neuropathy has been reported with medication use.
Pyruvate carboxylase deficiency
Pyruvate carboxylase (PC) is a nuclear-encoded mitochondrial enzyme that catalyzes the conversion of pyruvate to oxaloacetate. PC deficiency can be categorized into 3 types. Type A, found in North American Indians, involves lactic acidosis and psychomotor retardation. Type B, found in France and the United Kingdom, has a severe phenotype with hyperammonemia. Patients with type B die by age 3 months. [28] Type C manifests with relatively benign intermittent ataxia, and affected individuals may have normal development. PC deficiency usually manifests in the neonatal period with severe lactic acidosis or in early infancy with features similar to PDH deficiency with psychomotor retardation, hypotonia, and seizures.
-
Gene, inheritance, and pathogenesis: The most common disorder of pyruvate metabolism is an autosomal recessive inherited deficiency of PC. Identified mutations affect the gene locus on chromosome 11 (11q13.4-q13.5). Common founder 1828G-->A missense mutation has been described in Ojibway-Cree patients in Manitoba. [29]
-
Diagnosis
Lactic acidosis (elevated plasma lactate)
Increased lactate-to-pyruvate ratio
Elevated blood levels of ammonia, citrulline, proline, and lysine in type B (French form)
Reported abnormality on ultrastructural examination of skeletal muscle in the neonatal form: Subsarcolemmal aggregation of lipid droplets, glycogen granules, and pleomorphic mitochondria is found. Although nonspecific, these findings in combination with age of onset, clinical features, and lactic acidosis are often helpful in diagnosis.
Cystic periventricular white matter changes in the neonatal form on magnetic resonance imaging (MRI)
Assay for enzyme activity in cultured fibroblasts
Mutation testing
-
Treatment: Options are limited to symptomatic treatment of lactic acidosis and are similar to those employed for the treatment of PDH deficiency. Biotin and aspartate have been used in selected patients. Prognosis remains poor for types A and B.
Defects of mitochondrial fatty acid beta-oxidation
See the list below:
-
Gene, inheritance, and pathogenesis: Recessively inherited defects that affect mitochondrial beta-oxidation can result in intermittent episodes of neurologic symptoms (eg, weakness, ataxia, coma) in affected individuals. Defective fatty acid oxidation carries with it the consequence of energy deficit in the nervous system. The results are reflected in diffuse CNS dysfunction in situations of metabolic decompensation, such as that which accompanies prolonged fasting. Examples of such defects are as follows: [30]
Carnitine palmitoyltransferase-1 deficiency
Medium-chain acyl-CoA dehydrogenase deficiency (MCADD)
Multiple-acyl-CoA dehydrogenase deficiency (glutaric aciduria type II)
Primary systemic carnitine deficiency
Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency
Short-chain acyl-CoA dehydrogenase deficiency
Trifunctional enzyme deficiency
Very long-chain acyl-CoA dehydrogenase deficiency (VLCAD)
-
Clinical features
Episodic vomiting
Intermittent bouts of weakness, lethargy, ataxia, and coma
Neurologic symptoms induced by fasting
-
Diagnosis
Hypoglycemia with minimal-to-absent ketonemia and ketonuria
Mild lactic acidosis, hyperammonemia
Reduced plasma carnitine levels (free and total) in many fatty acid oxidation disorders
Increased dicarboxylic aciduria (suberic, sebacic, adipic acids) upon urinary organic acid analysis
Characteristic acylcarnitine profiles and urinary acyl-glycines associated with specific disorders of fatty acid oxidation
Specific enzyme assays on cultured skin fibroblasts
Mutation analysis (eg, the common A985G mutation in MCADD)
-
Treatment
Avoidance of prolonged fasting
Carnitine supplementation in doses of 50-100 mg/kg/d divided into 3 doses
Adequate caloric intake through intravenous glucose during acute presentations
Substitution of dietary fat with medium-chain triglycerides (may be helpful in bypassing metabolic block in VLCAD)
Corn starch feeds prior to bedtime (may help prevent hypoglycemia)
Urea cycle defects (late onset)
See the list below:
-
Gene, inheritance, and pathogenesis: Defects of each of the 5 enzymes of the urea cycle and 1 of its activators have been described. Most manifest with hyperammonemic coma in the neonatal period. Partial deficiencies can result in delayed presentation or intermittent symptoms during periods of decompensation. Elevated ammonia is poorly handled within the nervous system because of its ability to cross the blood-brain barrier. Secondary excitotoxicity related to release of glutamate and free radical–induced injury lead to diffuse cerebral dysfunction. Four of the 5 enzyme deficiencies (except ornithine transcarbamylase) are inherited as autosomal recessive defects. The 5 urea cycle enzymes are as follows: [31]
Carbamyl phosphate synthetase
Ornithine transcarbamylase (X-linked inheritance)
Argininosuccinate synthetase
Argininosuccinate lyase
Arginase
-
Clinical features: Delayed presentations of partial enzyme deficiencies in children and adults include the following:
Behavioral abnormalities such as self-abusive behavior
Episodic hyperammonemia
Intermittent ataxia and spasticity
Protein intolerance with intermittent vomiting
In adults, migrainelike episodes, confusional states, visual impairment, hallucinations, and neuropsychiatric symptoms
Presentation in ornithine transcarbamylase heterozygotes during pregnancy
Hyperactive deep tendon reflexes, papilledema, and decerebrate or decorticate posturing
Arginase deficiency clinically similar to spastic diplegic cerebral palsy [32]
-
Diagnosis [33]
Respiratory alkalosis
Elevated plasma ammonium (ionized form at physiologic pH)
Abnormalities in plasma amino acids
Elevated glutamine and alanine in blood and CSF
Indication of precise urea cycle enzyme deficiency possible by presence or absence of citrulline, argininosuccinic acid in plasma, and orotic acid in urine
Enzyme assays on tissue from liver biopsy
DNA analysis (can be confirmatory and is less invasive)
-
Treatment
Reduction of dietary protein intake with special dietary formulas
Supplementation of arginine and/or citrulline (depending on site of urea cycle defect)
Aggressive treatment of hyperammonemic coma using alternative pathway activation (eg, via sodium benzoate/sodium phenylacetate, and arginine)
Orthotopic liver transplant (another therapeutic option)
Gene therapy for OTC deficiency (remains experimental)
Table 3. Intermittent Ataxias Related to Enzyme Defects (Open Table in a new window)
Disorder/Syndrome |
Phenotype* |
Inheritance |
Locus/Gene |
Maple syrup urine disease |
Intermittent ataxia |
Autosomal recessive |
1p21.2 – DBT 6q14.1 – BCKDHB 19q13.2 – BCKDHA |
Hartnup disease |
Intermittent ataxia |
Autosomal recessive |
5p15.33 SLC6A19 |
Pyruvate dehydrogenase deficiency |
Intermittent ataxia Lactic acidosis |
X-linked recessive |
Xp22.12 |
Pyruvate carboxylase deficiency |
Intermittent ataxia Lactic acidosis |
Autosomal recessive |
11q13.2 PC |
Defects of mitochondrial fatty acid beta-oxidation |
Intermittent ataxia Metabolic acidosis Elevated ammonia |
Autosomal recessive |
N/A |
Late-onset urea cycle defects Argininosuccinic acidemia Carbamyl phosphate synthetase deficiency Citrullinemia Ornithine transcarbamoylase deficiency Argininemia |
Intermittent ataxia Episodic encephalopathy |
Autosomal recessive |
7q11.21 (arginosuccinate lyase) 2q34 (carbamoyl-phosphate synthetase I) 9q34.11 (arginosuccinate synthetase) Xp11.4 (ornithine carbamoyltransferase) 6q23.2 (arginase) |
Chronic or Progressive Ataxias
Inheritance factors
The following disorders are dominantly or recessively inherited. They manifest primarily with ataxia and cerebellar dysfunction, which are chronic and may be progressive with or without the presence of other neurologic abnormalities. This group of disorders is large; many have been associated with molecular genetic abnormalities, linking them to identifiable biochemical defects. DNA-based laboratory testing is available for many of these disorders. The salient phenotypic features and the responsible mutated genes are summarized in the tables accompanying this discussion.
Dominantly inherited ataxias
The number of dominantly inherited SCAs that have been described has increased to almost 50 and are labeled SCA1 onwards in sequence as they were discovered. The genetic basis for these disorders is varied. Some of them are related to expansion of triplet nucleotide repeats, which is most often a CAG repeat. A great degree of overlap in phenotype is noted, including the age of onset, with the major group of symptoms related to cerebellar and spinocerebellar pathway dysfunction. Other than distinguishing features described in selected cases, findings from neuroimaging studies are relatively nonspecific.
A slowly progressive cerebellar syndrome with various combinations of oculomotor disorders, dysarthria, dysmetria/kinetic tremor, and ataxic gait are key presenting features. In addition, pigmentary retinopathy, extrapyramidal movement disorders (parkinsonism, dyskinesias, dystonia, chorea), pyramidal signs, cortical symptoms (seizures, cognitive impairment/behavioral symptoms), and peripheral neuropathy are also noted.
The following selected clinical features are often helpful in predicting association with a gene defect:
-
SCA2 - Slowing of saccades
-
SCA1, SCA2, and SCA3 - Ophthalmoplegia
-
SCA1, SCA2, SCA3, SCA4, SCA8, SCA18, and SCA25 - Associated signs of peripheral neuropathy
-
SCA7 - Pigmentary retinopathy
-
SCA3 - Spasticity
-
SCA17 and DRPLA - Cognitive impairment/behavioral symptoms
-
SCA27 - Associated with dyskinesias
-
SCA10, SCA17, and DRPLA - Seizures
Three patterns of atrophy are described on brain MRI: pure cerebellar atrophy, olivopontocerebellar atrophy, and global brain atrophy. The presence of dentate nuclei calcifications in SCA20 can result in a hypointense/low signal on certain brain MRI sequences. Several identified mutations correspond to expansions of repeated trinucleotides (CAG repeats in SCA1, SCA2, SCA3, SCA6, SCA7, SCA-8, SCA-12, SCA17, and DRPLA; also CTG repeats in SCA8). A pentanucleotide repeat expansion (ATTCT) is associated with SCA10. A hexanucleotide repeat expansion (GGCCTG) is associated with SCA 36.
The following is a discussion of the clinical features of the dominantly inherited ataxias. Most of the SCAs are accounted for by the SCA1, SCA2, SCA3, SCA6, SCA7, and SCA8 subtypes; the remaining types are rare and have been reported in few families or in specific ethnic backgrounds. Treatment, for the most part, is restricted to the use of pharmacologic agents for targeted symptoms, such as the use of 5 hydroxytryptophan and acetazolamide for ataxia, amantadine/levodopa/dopamine agonists in SCA2-SCA3, and the use of tizanidine/baclofen for spasticity.
A clinical trial from has shown that varenicline (Chantix), can be used to improve axial symptoms and rapidly alternating movements in patients with SCA3. [34] However, other groups have failed to reproduce the same results. [35] Deep brain stimulation has been used for the treatment of tremor in SCA2.
Spinocerebellar ataxia 1
See the list below:
-
Autosomal dominant. CAG repeat expansion on chromosome 6p22.3 of the ATXN1 gene.
-
Clinical features
Onset in the third or fourth decade of life.
- Gait ataxia, incoordination, scanning speech, nystagmus, peripheral neuropathy, muscle wasting, and dystonia in late stages of the disease. Chorea may be present.
Spinocerebellar ataxia 2
See the list below:
-
Autosomal dominant. CAG repeat expansion on chromosome 12q24.12of the ATXN2 gene.
-
Clinical features
The age of onset is during infancy, but adult onset form has been reported.
Childhood onset is associated with hypotonia, infantile spasm, autonomic dysfunction, dysphagia, and retinitis pigmentosa. Adult onset is associated with slow saccades, ataxia, and hyporeflexia. Developmental delay, intellectual impairment, and dementia can also be seen.
Spinocerebellar ataxia 3
The disorder is also known as Machado-Joseph disease (MJD). It was originally described as affecting individuals of Portuguese-Azorean descent. There are recognized clinical subtypes of (MJD) (37). This condition has also been found in other populations, including German, Africa, and Chinese (116, 117, 118).
-
Autosomal dominant. CAG expansions in the chromosome 14q32 of ATXN3 gene.
-
This is the most common form of spinal cerebellar ataxia affecting approximately 21% of the SCA in the United States.
-
Clinical features
Age of onset after the fourth decade of life. The earlier onset of the disease is associated with a more severe form of the disease.
The main symptoms are cerebellar signs, ophthalmoplegia, pyramidal signs, and extrapyramidal signs.
Type 1 has an early onset of pyramidal symptoms and dystonia. Type 2 has pure cerebellar ataxia. Type 3 has later onset and peripheral neuropathy. However, many people have overlapping symptoms so classifying into subtypes may not be useful clinically.
Ataxia, spasticity, ophthalmoplegia, fasciculation, nystagmus, pyramidal and extrapyramidal signs, amyotrophy.
Spinocerebellar ataxia 4
See the list below:
-
Autosomal dominant. CAG repeats expansion on chromosome 6q22.1. The gene mutation has not been identified.
-
Clinical features
Onset has been documented as early as the second decade, but more commonly it is in the third or fourth decade of life.
Symptoms include gait difficulty, ataxia, dysarthria, dysmetria, and axonal sensory neuropathy. Hyporeflexia is documented.
Spinocerebellar ataxia 5
See the list below:
-
Autosomal dominant. Mutation in the chromosome 11p13 of the SPTBN2 gene.
-
Clinical features
Age of onset variable, with a mean age of 37 years (10–68 y). Anticipation is documented.
Cerebellar ataxia, facial myokymia, dysmetria, downbeat nystagmus, and very slow progression
First family described descending from Abraham Lincoln's grandparents; second family described in northeastern France
Spinocerebellar ataxia 6
See the list below:
-
Autosomal dominant. CAG repeat in chromosome 19p13 of the CACNA1A gene.
CACNA1A is also associated with episodic ataxia type 2 and familial hemiplegic migraine.
-
Clinical features
Symptoms beginning in the fourth or fifth decade of life.
Slow progression over 20-30 years. Sometimes it can take patients a long time to notice they even have a problem due to insidious onset.
Patients develop ataxia, coordination difficulties, nystagmus, dysarthria, and loss of vibration and joint position sense and eventually become wheelchair bound. In a few patients with advanced age, chocking was observed.(9)
Spinocerebellar ataxia 7
See the list below:
-
Autosomal dominant. Glutamine repeat on chromosome 3p14 of the ATXN 7 gene.
-
Clinical features
Onset ranges from infancy (rare) to adulthood, with genetic anticipation observed.
Macular degeneration leading to vision loss is a unique feature to SCA 7. Retinal degeneration has also been reported.
Other symptoms include progressive ataxia and variable ophthalmoplegia, dysarthria, pyramidal and extrapyramidal signs, and impaired vibration sense. Childhood onset is associated with myoclonic seizures, vision loss, and cardiac problems.
Spinocerebellar ataxia 8
See the list below:
-
Autosomal dominant. A trinucleotide CTG repeat expansion on chromosome 13q21 of the ATXN9OS gene as well as trinucleotide CAG repeat expansion of the ATXN8 gene.
-
Clinical features
Onset of symptoms ranging from age 18-65 years, with a mean of 39 years.
Dysarthria, gait instability, and mild aspiration are commonly the initial symptoms. Other symptoms include nystagmus, spasticity, and diminished vibration perception. Progression is generally slow. People who are severely affected are wheelchair bound by fourth to fifth decade.
Disease severity correlates with length of trinucleotide repeat and patient age. (12)
Spinocerebellar ataxia 9
See the list below:
-
Unclear genetics at this time, but is passed along in autosomal dominant fashion.
-
Clinical features
Adult onset.
Presenting symptoms include imbalance and ataxia.
Variable symptoms include ophthalmoplegia, dysarthria, pyramidal and extrapyramidal tract signs, weakness, posterior column signs. Two patients had parkinsonism, one patient had presentation resembling multiple sclerosis, including demyelinating brain lesions on MRI brain.
Disease was found in family with British ancestry.(29)
Spinocerebellar ataxia 10
See the list below:
-
Autosomal dominant. ATTCT pentanucleotide repeats in the chromosome 22q13.31of the ATXN10 gene.
-
Clinical features
Onset in third to fifth decade of life.
- All patients have gait ataxia, dysarthria, dysmetria, dysdiadochokinesis, nystagmus. Some patients also had seizures, dysphagia or dementia. (13)(14)
Spinocerebellar ataxia 11
See the list below:
-
Autosomal dominant. Mutation in the chromosome 15q15.2 of the TTBK2 gene.
-
Clinical features
Normal life span with mean age of onset of 30 years (15–70 y).
Mild disorder, with pure ataxia as a major feature. Retained capacity for ambulation.
Spinocerebellar ataxia 12
See the list below:
-
Autosomal dominant. CAG repeats expansion on chromosome 5q31-5q32of the PPP2R2B gene.
-
Clinical features
Onset is in the fourth decade.
Action tremor is the first presenting sign. Patients then develop ataxia and other cerebellar signs.
Patients have hyperreflexia, abnormal eye movements, and dementia found in the oldest patients
Beta blocker and benzodiazepines can sometimes improve tremor. (15)
Spinocerebellar ataxia 13
See the list below:
-
Autosomal dominant. Mutation in the chromosome 19q13 of the KCNC3 gene.
-
Clinical features
Onset can be childhood or older.
Presents with cerebellar gait ataxia. Associated dysarthria, intellectual disability, motor development delay, nystagmus, and pyramidal signs. Slow progression of symptoms.(16)
Spinocerebellar ataxia 14
See the list below:
-
Autosomal dominant. Mutation in chromosome 19q13.4 of the PRKCG gene.
-
Clinical features
Onset mean age was 40 years, some with early onset.
Patients with early onset first presented with axial myoclonus followed by progressive ataxia. In patients with later onset, gait disorder is usually the presenting feature.
Variable other symptoms include cerebellar dysarthria, slow saccades, ocular dysmetria, and hyperreflexia. (17, Yamashita) (18, van de Warrenburg).
Spinocerebellar ataxia 15/16
The diagnosis of SCA16 has now been included into the diagnosis of SCA15. [36]
-
Autosomal dominant. Mutation in the chromosome 3p26 of the ITPR1 gene.
-
Clinical features
Adult onset.
Patients develop slowly progressive cerebellar ataxia. Most patients have severe disabling action and postural tremor. Other variable symptoms include gaze palsy, pyramidal tract signs, and dorsal column involvement. Patients also have mild cognitive dysfunction. (19)
Spinocerebellar ataxia 17
See the list below:
-
Autosomal dominant. CAG repeats in the chromosome 6q27 in the TBP gene.
-
Clinical features
Age of onset ranges from childhood to adult.
Symptoms include ataxia, pyramidal and extrapyramidal signs, cognitive impairment, psychosis and seizures. Patients also had hyperreflexia.
Some patients have presentations that are indistinguishable from Huntington disease. (17) (21)
Spinocerebellar ataxia 18
See the list below:
-
Autosomal dominant. Mutation in the chromosome 7q22-q32. The gene mutation has not been identified.
-
Clinical features
Age of onset in the second and third decade of life.
Gait difficulty was the most common initial symptom.
Other symptoms include dysmetria, decreased vibratory and proprioceptive sense, muscle weakness and atrophy. Several patients had pes cavus.
All patients were of Irish ancestry. (28)
Spinocerebellar ataxia 19/22
SCA 19 is also known as SCA 22.
-
Autosomal dominant. Mutation in the chromosome 1p13.2 on gene KCND3.
-
Clinical features
The age of onset is in the third decade.
Mild ataxia syndrome with cognitive impairment, myoclonus, and postural tremor.
Some had dysphagia, dysarthria, or nystagmus, impaired vibration, mild cogwheel rigidity, urinary urgency or incontinences. (22), (23)
Spinocerebellar ataxia 20
See the list below:
-
Autosomal dominant. Mutation in the chromosome 11q12.2-11q12.3, overlapping with SCA5.
-
Clinical features
Age of onset - 19–64 years (median 46.5 years).
Most common symptom is dysarthria due to spasmodic dysphonia, followed by gait ataxia and upper limb ataxia.
Slow progression of disease. Patient’s rarely become wheelchair bound.
Other symptoms which were variable include mild pyramidal signs, hypermetric saccades, nystagmus, spasmodic coughing. (24) (25)
Spinocerebellar ataxia 21
See the list below:
-
Autosomal dominant. Mutation in the chromosome 1p36 of the TMEM240 gene.
-
Clinical features
Age of onset usually between 1 and 30 years, although some presented between 40 and 61 years of age.
Symptoms include gait and limb ataxia. Cognitive impairment is very common.
Other variable symptoms include akinesia, dysarthria, dysgraphia, microsaccadic pursuit, square wave jerks, extrapyramidal signs (tremor, parkinsonism [not responsive to L-DOPA], cogwheel rigidity). (26) (27)
Spinocerebellar ataxia 23
See the list below:
-
Autosomal dominant. Mutation in the chromosome 20p13 of PDYN gene.
-
Clinical features
Late onset, range between 43 and 56 years of age.
Presenting symptoms include slowly progressive gait and limb ataxia.
Other variable symptoms include dysarthria, ocular dysmetria, slow saccades, and decreased vibratory sense below the knees. Some patients had tremor and memory deficits that began around the age of 50 that could progress to dementia. Some patients had hyperreflexia.
Patients rarely became wheelchair bound. (30) (31)
Spinocerebellar ataxia 25
See the list below:
-
Autosomal dominant. Mutation in the chromosome 2p21-p13.
-
Clinical features
Age of onset ranges between 17 months and 39 years, although childhood onset is more common.
Patients all had cerebellar ataxia, to varying degrees.
Many patients had areflexia in lower limbs and had a peripheral sensory neuropathy.
Other variable features include nystagmus, decreased visual acuity facial tics, urinary urgency and gastrointestinal symptoms. (32)
Spinocerebellar ataxia 26
See the list below:
-
Autosomal dominant. Mutation in the chromosome 19p13.3 of the EEF2 gene.
-
Clinical features
Age of onset ranges between 26 and 60 years (mean age is 42 y)
Symptoms include ataxia of trunk and limbs, dysarthria, and irregular visual pursuit.
Disease was slowly progressive. (33)
Spinocerebellar ataxia 27
See the list below:
-
Autosomal dominant. Mutation in the chromosome 13p33 of the FGF14 gene.
-
Clinical features
Early disease onset.
First symptoms are tremor in childhood. Ataxia usually develops in the second decade. Most patients had intellectual disability and aggressive outbursts.
Variable symptoms include nystagmus, cerebellar dysarthria, orofacial dyskinesias, severe limb ataxia, red-green colorblindness, strabismus, and inability to complete school.
Initially described in a Dutch family. (34) (35)
Spinocerebellar ataxia 28
See the list below:
-
Autosomal dominant. Mutation in the chromosome 18p11 of the AFG3L2 gene.
-
Clinical features
Age of onset ranges between 6 and 60 years (mean age 30.7 y).
Slowly progressive and does not result in major functional incapacity.
Patients usually present with cerebellar ataxia.
Variable symptoms include dysarthria, ophthalmoplegia, nystagmus, saccadic pursuit, ptosis, pyramidal symptoms, spasticity.
Disease has been described in families with Italian and French origin. (36)
Spinocerebellar ataxia 29
See the list below:
-
Autosomal dominant. Mutation in the chromosome 3p26 of the ITPR1 gene.
-
Clinical features
Usually congenital or early childhood onset
Patients have nonprogressive ataxia, wide based gait. Dysarthria, cognitive impairment, and frequent falling are common additional features.
Other variable symptoms include nystagmus and dysdiadochokinesis.
Some patients had improvement in motor symptoms with increasing age. (37, 38, 39)
Spinocerebellar ataxia 30
See the list below:
-
Autosomal dominant. Mutation in the chromosome 4q34.3-q35.1.
-
Clinical features
Age of onset ranges between 42 and 76 years of age (mean age 52 y).
Relatively pure, slowly progressing gait and appendicular ataxia with mild to moderate dysarthria.
Some patients had lower limb hyperreflexia and nystagmus. (40)
Spinocerebellar ataxia 31
See the list below:
-
Autosomal dominant. Mutation in the chromosome 16q21 of the BEAN gene.
-
Clinical features
Some patients had hearing loss. (41, 42)
Symptoms include gait ataxia, nystagmus, cerebellar dysarthria, limb ataxia decreased muscle tone.
Late age of onset (on average at approximately 60 years of age).
Spinocerebellar ataxia 32
See the list below:
-
Autosomal dominant. Mutation in the chromosome 7q32-q33.
-
Clinical features
Broad age of onset.
People with age of onset before 40 had cognitive impairment and cerebellar atrophy.
Males are infertile with azoospermia and testicular atrophy.
Spinocerebellar ataxia 34
See the list below:
-
Autosomal dominant. Mutation in the chromosome 6q14 of the ELOVL4 gene.
See the list below:
-
Clinical features
Onset of ataxia is usually around 40 years of age.
Some patients have skin lesions that develop soon after birth and usually resolve by the age of 25. The skin lesions are characterized as papulosquamous erythematous ichthyosiform plaques.
Other variable symptoms include hyporeflexia, nystagmus, dysarthria, supranuclear ophthalmoplegia, and autonomic symptoms.
Ataxia can be severe and many patients become wheelchair bound late in life.
Spinocerebellar ataxia 35
See the list below:
-
Autosomal dominant. Mutation in the chromosome 20p13 of the TGM6 gene.
-
Clinical features
Most become wheelchair bound after 10 years.
Additional variable features include tremor, torticollis, ocular dysmetria and position sense defects.
Early features are difficulty walking, ataxia, cerebellar dysarthria. Later features are upper limb incoordination.
Age of onset ranges from 40 to 48 years of age (mean is 43.9 years).
Spinocerebellar ataxia 36
See the list below:
-
Autosomal Dominant. GGCCTG hexanucleotide repeats in the chromosome 20p13 of the NOP56 gene.
-
Clinical features
Variable features include tongue atrophy or fasciculation, dysphagia, eye movement abnormalities, hearing loss.
Symptoms include gait ataxia, truncal instability, dysarthria and limb incoordination.
Age of onset is on average 53 years of age.
Spinocerebellar ataxia 37
See the list below:
-
Autosomal dominant. Mutation in the chromosome 1p32 of the DAB1 gene.
-
Clinical features
The age of onset is in the fourth decade of life.
Initial symptoms include increased falls, gait instability, dysarthria, clumsiness. Most patients had dysmetric vertical saccades and irregular vertical pursuit.
Variable features include trunk ataxia, dysmetria, and dysphagia, tremor, oscillopsia, nystagmus.
Slow clinical progress, some becoming wheelchair bound (43).
Spinocerebellar ataxia 38
See the list below:
-
Autosomal dominant. Mutation in the chromosome 6p12 of the ELOVL5 gene.
-
Clinical features
The age of onset is between third to fifth decade of life.
Presenting symptoms include walking difficulties.
Other variable symptoms include nystagmus, slow saccades, dysarthria, and limb ataxia, and axonal neuropathy (44).
Spinocerebellar ataxia 39
See the list below:
-
Mutation in 11q21-11q22.3
-
Clinical features
Symptoms include strabismus, saccadic pursuit dysfunction, horizontal gaze palsy, and mild intellectual disability (45).
Spinocerebellar ataxia 40
See the list below:
-
Autosomal dominant. Mutation in the chromosome 14q32 of the CCDC88C gene.
-
Clinical features
Adult onset.
Presenting symptoms are ataxic gait and dysarthria.
Other features include wide-based gait, ocular dysmetria, intention tremor, scanning speech, impaired vertical gaze.
Some patients become wheelchair bound about 17 years after presenting symptoms (46).
Spinocerebellar ataxia 41
See the list below:
-
Autosomal dominant. Mutation in the chromosome 14q32 of the CCDC88C gene.
-
Clinical features
Adult onset.
Presenting symptoms were progressive imbalance and gait ataxia (47).
Spinocerebellar ataxia 42
See the list below:
-
Autosomal dominant. Mutation in the chromosome 17q21 of the CACNA1G gene.
-
Clinical features
Highly variable age of onset (on average is mid-adult) and disease severity.
Presenting symptoms gait instability.
Variable features include dysarthria, saccadic eye movements, diplopia, and nystagmus. Less common features include decreased vibratory sense, spasticity, and urinary dysfunction.
Progression of disease is relatively slow (48).
Spinocerebellar ataxia 43
See the list below:
-
Autosomal dominant. Mutation in the chromosome 3q25 of the MME gene.
-
Clinical features
Adult onset.
Presenting symptoms include slowly progressive gait and limb ataxia. It is often associated with peripheral neuropathy.
Variable features include pes cavus, mild atrophy of lower limbs, mild cogwheel rigidity, hypometric saccades, tremor, nystagmus, and dysarthria (49).
Spinocerebellar ataxia 44
See the list below:
-
Autosomal dominant. Mutation in the chromosome 6q24 of the GRM1 gene.
-
Clinical features
Adult onset usually, although can range between 5 years of age (rarely) to 50 years of age.
Presenting features include gait ataxia, frequent falls.
Variable features include dysarthria, dysphagia, dysmetria and dysdiadochokinesis.
Not usually wheelchair bound or severely disabled (50).
Spinocerebellar ataxia 45
See the list below:
-
Autosomal dominant. Mutation in the chromosome 5q33 of the FAT2 gene.
-
Clinical features
Adult onset.
Symptoms are relatively pure cerebellar syndrome, including limb and gait ataxia, downbeat nystagmus and dysarthria (51).
Spinocerebellar ataxia 46
See the list below:
-
Autosomal dominant. Mutation in the chromosome 5q33 of the FAT2 gene.
-
Clinical features
Adult onset (average is 53 years of age).
Symptoms are sensory neuropathy and cerebellar ataxia.
Variable features include cerebellar dysarthria. Patient rarely have abnormal oculomotor function (52).
Spinocerebellar ataxia 47
See the list below:
-
Autosomal dominant. Mutation in the chromosome 1p35 of the PUM1 gene.
-
Clinical features
Onset in third or fourth decade.
Presenting symptoms are slowly progressive gait ataxia, dysmetria, dysarthria. Some patients had diplopia.
There is an early onset form also reported. Features include delayed motor development, early onset ataxia, short stature. Some patients had chorea, dysarthria, spasticity, ballismus, seizures, facial dysmorphism, and incoordination (53).
Spinocerebellar ataxia 48
See the list below:
-
Autosomal dominant. Mutation in the chromosome 16p13 of the STUB1 gene.
-
Clinical features
Adult onset (median age of 42 years).
Features are progressive cognitive decline with associated ataxia.
Roughly half of patients have cognitive or affective dysfunction that proceeds onset of ataxia. The other half of patients have ataxia then develop cognitive or affective problems.
Examples of cognitive and affective impairment includes anxiety, agoraphobia, declines in cognitive or executive function.
Motor symptoms include ataxia, dysarthria, dysphagia, ocular dysmetria, urinary incontinence. Some patients become wheelchair bound (54).
Dentatorubro-pallidoluysian atrophy (DRPLA)
See the list below:
-
Gene, inheritance, and pathogenesis: Autosomal dominant, caused by a trinucleotide repeat (CAG) in the ATN1 gene on chromosome 12p. More common in the Japanese population. However, there is a condition that is allelic to the Haw River syndrome reported in African-American descendants in North Carolina.
- Pathologic features include nerve cell loss and gliosis affecting the dentate nucleus, red nucleus, pallidum, and subthalamic nucleus of Luys. The age of onset varies; can be childhood but usually it is in the twenties with death in the forties.
- DRPLA gene is also known as atrophin-1, a transcription factor located in the nucleus. Mutation in the DRPLA gene causes pathological accumulation of atrophin-1 in the neuronal nuclei causing central nervous system dysfunction.
-
There are 3 clinical forms of DRPLA identified: the ataxo-choreoathetoid form, the peudo-Huntington form, and the myoclonic epilepsy form.
-
Clinical features
- Ataxia
- Dementia
- Polymyoclonus
- Chorea
- Myoclonic epilepsy
-
The disease has an anticipatory nature. If the disease appears before the age of 20, it is characterized by myoclonus epilepsy, intellectual disability, behavioral problems, and ataxia. If the disease present after the age of 20, it is characterized by choreoathetosis, ataxia, psychiatric symptoms, and dementia.
-
Diagnosis
- Imaging studies demonstrate spinocerebellar atrophy and varying degrees of multisystem atrophy.
- Diagnosis rests on molecular DNA confirmation of expansion of the number of CAG repeats.(55, 56).
-
Treatment
- A multidisciplinary approach and mainly supportive therapy.
- Physical therapy and occupational therapy for ataxia.
- Tetrabenazine, risperidone, gabapentin for choreoathetoid and dystonic movement.
- Antiepileptic medication for seizure.
- Genetic counseling for the family members as the disease has an autosomal dominant pattern with anticipation.
- Palliative care.
- Gene replacement therapy with adenovirus vector and intrathecal antisense oligonucleotide (ASO) are under investigation.
Cerebellar ataxia nonprogressive, with mental retardation (CANPMR)
See the list below:
-
Gene, inheritance, and pathogenesis: Autosomal dominant. Deletion of the CAMPTA1 gene, causing a frameshift and premature termination.
-
Clinical features
- Symptoms apparent in infancy.
- Ataxia with associated intellectual disability.
- Variable features include hypotonia, dysarthria, dysmorphic facial features, myoclonic seizures (57).
Cerebellar ataxia, deafness and narcolepsy, autosomal dominant (ADCADN)
See the list below:
-
Gene, inheritance, and pathogenesis: Autosomal dominant, caused by a mutation of DNMT1 gene.
-
Clinical features
- Cerebellar ataxia, narcolepsy/cataplexy, sensorineural deafness and dementia.
- Variable features include optic atrophy and psychiatric symptoms, tremor (58).
Table 4. Dominantly Inherited Chronic/Progressive Ataxias (Open Table in a new window)
| Autosomal Dominant Ataxias | Neurologic Phenotype (Gait ataxia is a constant feature) |
Locus/Gene |
|---|---|---|
| SCA1 | Peripheral neuropathy Pyramidal signs Ophthalmoparesis |
6p22.3 ATXN1 |
| SCA2 | Slow saccades Facial fasciculation Hyporeflexia Dementia Peripheral neuropathy Extrapyramidal findings |
12q24.12 ATXN2 |
| SCA3 | Pyramidal and extrapyramidal signs Opthalmoplegia Eyelid retraction Amyotrophy Peripheral neuropathy |
14q32.12 ATXN3 |
| SCA4 | Sensory axonal neuropathy Pyramidal signs |
16q22.1 |
| SCA5 | Early onset, relatively pure cerebellar ataxia with dysarthria Slow progression |
11p13.2 SPTBN2 |
| SCA6 | Slow onset pure cerebellar ataxia with dysarthria, nystagmus | 19p13.13 CACNA1A |
| SCA7 | Vision loss - macular or retinal degeneration Dysarthria |
3p14.1 ATXN7 |
| SCA8 | Hyperreflexia, spasticity Impaired vibration sense |
13q21 ATXN8 |
| SCA9 | Ophthalmoplegia Dysarthria Pyramidal and extrapyramidal tract signs Weakness Posterior column signs |
Unknown |
| SCA10 | Pure cerebellar syndrome Seizures Dementia and dysphagia rarely |
22q13.31 ATXN10 |
| SCA11 | Slowly progressive mild ataxia |
15q15.2 TTBK2 |
| SCA12 | Tremor at onset Hyperreflexia Late dementia |
5q32 PPP2R2B |
| SCA13 | Childhood onset Associated cognitive and motor delays |
19q13.33 KCNC3 |
| SCA14 | Axial myoclonus Eye movement abnormalities |
19q13.42 PRKCG |
SCA15/SCA 16 |
Pure ataxia with slow progression Postural tremor Gaze palsy |
3p26.1 ITPR1 |
SCA17 |
Cognitive impairment Psychosis Seizures Huntington disease-like presentation Chorea |
6q27 TBP |
SCA18 |
Dysmetria Sensory axonal neuropathy Muscle weakness & atrophy Pes cavus |
7q22-q32 |
SCA19/22 |
Slowly progressive ataxia Hyporeflexia Cognitive decline Myoclonus |
1p13.2 KCND3 |
SCA20 |
Dysarthria and dysphonia Palatal tremor Spasmodic coughing |
11q12 |
| SCA21 | Cognitive disorders Extrapyramidal features Dysarthria Can have childhood onset |
1p36.33 TMEM240 |
SCA23 |
Sensory loss Vibration loss Tremor Memory deficits after 50 |
20p13 PDYN |
| SCA25 | Childhood onset most common Sensory neuropathy Gastrointestinal symptoms Nystagmus Facial tics |
2p21-p13 |
| SCA26 | Dysarthria Ocular pursuit abnormalities |
19p13.3 EEF2 |
SCA27 |
Tremor then gait and limb ataxia Behavioral outbursts Red-green colorblindness Strabismus |
13q33.1 FGF14 |
| SCA28 | Ophthalmoparesis Dysarthria Nystagmus Ptosis |
18p11.21 AFG3L2 |
| SCA29 | Congenital or childhood onset Nonprogressive ataxia Cognitive impairment Frequent falls Some motor symptoms can improve with age |
3p26.1 ITPR1 |
| SCA30 | Pure cerebellar syndrome Dysarthria Lower limb hyperreflexia |
4q34.3-q35.1 |
SCA31 |
Slowly progressive gait and limb ataxia Hearing loss Cerebellar dysarthria |
16q21 BEAN |
SCA32 |
Variable cognitive impairment, and azoospermia in males | 7q32-q33 |
| SCA34 | Papulosquamous erythematous plaques | 6q14.1 ELOVL4 |
| SCA35 | Upper limb involvement Torticollis |
20p13 TGM6 |
| SCA36 | Truncal instability Dysarthria Dysphagia Tongue atrophy or fasciculation Eye movement abnormalities Hearing loss |
20p13 NOP56 |
| SCA37 | Late onset falls and clumsiness Dysarthria Abnormal vertical eye movements |
1p32.2 DAB1 |
| SCA38 | Polyneuropathy Nystagmus Slow saccades |
6p12.1 ELOVL5 |
| SCA39 | Strabismus Saccadic pursuit dysfunction Horizontal gaze palsy Mild intellectual disability |
11q21-11q22.3 |
| SCA40 | Dysarthria Ocular dysmetria Tremor |
14q32.11-q32.12 CCDC88C |
| SCA41 | Late onset imbalance and gait ataxia | 4q27 TRPC3 |
| SCA42 | Dysarthria Saccadic eye movements Diplopia Nystagmus Spasticity |
17q21.33 CACNA1G |
| SCA43 | Slowly progressive gait and limb ataxia Peripheral neuropathy Mild atrophy of lower limbs |
3q25.2 MME |
| SCA44 | Frequent falls Dysarthria Dysphagia Dysmetria |
6q24.3 GRM1 |
| SCA45 | Relatively pure cerebellar syndrome Downbeat nystagmus Dysarthria |
5q33.1 FAT2 |
| SCA46 | Sensory neuropathy Cerebellar dysarthria |
19q13.2 PLD3 |
| SCA47 | Dysmetria Dysarthria Diplopia |
1p35.2 PUM1 |
| SCA48 | Cerebellar signs Cognitive impairment Anxiety |
16p13.3 STUB1 |
| CANPMR | Intellectual disability Hypotonia Dysarthria Dysmorphic facial features |
1p36.31-p36.23 CAMTA1 |
| ADCADN | Narcolepsy Sensorineural deafness Dementia |
19p13.2 DNMT1 |
| Dentatorubropallidoluysian atrophy (DRPLA) | Chorea Seizures Myoclonus Dementia |
12p13.31 ATN1 |
| *Gait ataxia is a constant feature |
Recessively inherited ataxias with spinocerebellar dysfunction
Recessively inherited ataxias with spinocerebellar dysfunction are discussed below.
Ataxia with selective vitamin E deficiency
See the list below:
-
Gene, inheritance, and pathogenesis: This is a rare autosomal recessive disorder resulting from a mutation that affects the gene for alpha-tocopherol transfer protein.
-
Clinical features
It is phenotypically similar to Friedreich ataxia (FRDA), with head titubation (28%), SCA, areflexia, and proprioception loss.
Skin is affected by xanthelasmata and tendon xanthomas.
Onset varies from ages 2 to 52 years and usually occurs in people younger than 20 years; it slowly progresses over decades.
-
Diagnosis: Measurements include low-to-absent serum vitamin E and high serum cholesterol, triglyceride, and beta-lipoprotein.
Friedreich ataxia
See the list below:
-
Gene, inheritance, and pathogenesis
- Friedreich’s Ataxia (FA) is an autosomal recessive ataxia. It is also the most common cause of ataxia.
- The disease is caused by a gene mutation of frataxin (FXN) on chromosome 9q13 due to GAA repeats on both alleles (61). However, patients with atypical presentations of FA are due to compound heterozygous mutations, meaning one allele has a GAA mutation and the other allele has a non-GAA mutation (62).
- The length of the GAA repeat correlates with the earlier onset and severity of the disease (63). In FA patients, 66-17000 GAA repeats are seen. If the repeat is interrupted by other types of non-GAA repeat mutations, it stabilizes the gene. If the gene goes uninterrupted, it causes expansion of over 300 genes in a single generation (64).
- GAA repeats impair the function of Frataxin, which is a mitochondria protein that promotes iron chaperoning, iron detoxication, antioxidation, and iron sulfur cluster biogenesis. It is highly expressed in the brain, heart, and pancreas (65).
-
Clinical features
- Onset – Typically adolescence, or generally under the age of 25.
- Neurological symptoms
- Up to 90% have cerebellar involvement including ataxic gait, uncoordinated movement, nystagmus, cerebellar dysarthria, and dysphagia.
- Up to 90% have dorsal root and peripheral nerve involvement involving sensory axonal neuropathy, proprioception impairment, vibration impairment, and diminished deep tendon reflexes.
- Motor weakness in the feet and legs can be seen first, followed by arms.
- Up to 50% have bladder dysfunction.
- Less than 30% have decrease in visual acuity, optic atrophy, or hearing loss.
- Cognition is preserved.
- Cardiac findings –Symmetric concentric hypertrophic cardiomyopathy.
- Skeletal findings – Scoliosis and pes cavus.
- Metabolic abnormalities – diabetes mellitus.
- Patients with atypical presentation due to compound heterozygous mutation have later onset (age over 25 years old), increased deep tendon reflexes, and spasticity.
-
Diagnosis
- Genetic testing for triple repeat expansion of FXN gene.
- Frataxin levels in blood or buccal cells using immunoassay. This can be useful for pre-symptomatic carriers or patients whose genetic testing did not show pathogenic mutations. In some clinics, immunoassay is the standard testing for FA (66).
- Abnormal electrocardiographic or echocardiographic findings are supportive features.
- Abnormal findings on motor and sensory nerve conduction studies are supportive features.
- MRI of the brain shows absence of cerebellar atrophy. Cerebellar atrophy does not rule out FA but one must consider other forms of hereditary ataxia as the diagnosis (67).
-
Treatment
- There is no cure or disease modifying treatment at this point.
- Supportive treatment including physical therapy and occupational therapy.
- Routine dysphagia screen, scoliosis screen, ophthalmology evaluation, audiology evaluation, and diabetes screening are important.
- Gene replacement therapy is under investigation.
- Omaveloxolone, an Nrf2 activator, is showing promising results. Omaveloxolone increases the level of Nrf2 and results in trigging of anti-inflammation cascades (68).
- Riluzole is shown to improve ataxia in a mixed group of patients including FA and other types of hereditary ataxia.
- Several anti-oxidants treatments such as Idebenone, Coenzyme Q10, Vitamin E, Resveratrol, A001, and L-Carnitine showed no clinical improvement. Nicotinamide, interferon gamma, and erythropoietin did not show clinical improvement as well. Iron chelation with deferiprone showed worsened outcomes (69).
Abetalipoproteinemia
See the list below:
-
Gene, inheritance, and pathogenesis: This rare autosomal recessive disorder is characterized by hypocholesterolemia and malabsorption of lipid-soluble vitamins. It features defective assembly and secretion of apolipoprotein B (Apo-B)–containing lipoproteins by the intestines and the liver. Mutations appear to affect the microsomal triglyceride transfer protein (MTP) gene. The heterodimeric protein is responsible for transfer of neutral lipids across cell membranes.
-
Clinical features
- Pigmentary degeneration of the retina
- Progressive ataxia
- Peripheral neuropathy
- Malabsorptive state in the early years with steatorrhea and abdominal distension
-
Diagnosis
- Acanthocytosis on peripheral blood smears
- Decreased serum cholesterol
- Serum beta lipoprotein absent
Hypobetalipoproteinemia
Clinically similar to abetalipoproteinemia, but can be more mild. It is also an autosomal codominat disorder. It is clinically indistinguishable from abetalipoproteinemia in its homozygous form. It is caused by mutations that affect the APOB gene, which affects turnover of Apo-B. Neurologic and nonneurologic manifestations are similar in homozygotes. Heterozygotes, on occasion, also may be affected. It is characterized by extremely low plasma levels of Apo-B as well as low levels of total cholesterol and LDL cholesterol. [70]
Table 5. Recessively Inherited Chronic/Progressive Ataxias with Spinocerebellar Dysfunction (Open Table in a new window)
| Disorder/Syndrome
|
Neurologic Phenotype | Inheritance | Locus/ Gene |
| Ataxia with selective vitamin E deficiency | Chronic ataxia | Autosomal recessive | 8q12.3 TTPA |
| Friedreich ataxia | Progressive ataxia plus | Autosomal recessive | 9q13-q21.11 FXN |
| Abetalipoproteinemia | Retinal degeneration Progressive ataxia Peripheral neuropathy |
Autosomal recessive | 4q24 MTTP |
| Hypobetalipoproteinemia | Retinal degeneration Progressive ataxia Peripheral neuropathy |
Autosomal codominant | 2q24 APOB |
| *Listed here due to overlap of clinical features with abetalipoproteinemia. | |||
Recessively inherited ataxias associated with defective DNA repair
The disorders discussed below involve defects in the DNA repair pathway. This is a complex pathway and breakdown at any point can have serious consequences, including increased risk of cancer, increased rate of aging, and a myriad of other conditions. Many genes affected below are critical to maintaining cellular resistance to ionizing radiation. Due to the increased risk of cancer and no specific curative treatments, many of these diseases carry a poor prognosis. [71]
Cockayne syndrome
See the list below:
-
Gene, inheritance, and pathogenesis: Type 1 (or A) and type II (or B) are the 2 predominant forms. Inheritance is autosomal recessive for both. Defective repair of transcriptionally active DNA is the underlying basis of the disorder. Cultured skin fibroblasts from these patients display abnormal UV sensitivity. Mutations in the excision-repair cross-complementing group 8 gene (ERCC8) in type I or the excision-repair cross-complementing group 6 gene (ERCC6) in type II result in Cockayne syndrome. Early death in the second or third decade is usual.
-
Clinical features
- Failure to thrive, growth failure, dwarfism
- Sensorineural hearing loss
- Microcephalic and distinctive facies
- Intellectual disability
- Clinical photosensitivity
- Persistently cold hands and feet
- Other variable features include gait disturbances, congenital cataracts, loss of adipose tissue, joint contractures, pigmentary retinopathy
- No increase in incidence of malignancy in these patients
De Sanctis-Cacchione
See the list below:
-
Gene, inheritance, and pathogenesis
- This autosomal recessive condition can be seen with several different forms of xeroderma pigmentosum. It is termed a “xerodermic idiocy,” where a patient has the symptoms of xeroderma pigmentosum, intellectual disability, progressive neurologic deterioration, dwarfism, and gonadal hypoplasia.
- Xeroderma pigmentosum is due to a defect in DNA excision repair following UV exposure. There is a mutation in gene ERCC6.
-
Clinical features
- Cutaneous photosensitivity and multiple cancers
- Mental and motor retardation
- Progressive neurologic decline
- Peripheral sensory neuropathy
-
Diagnosis: Defective DNA repair after ultraviolet radiation damage [73]
Ataxia telangiectasia
See the list below:
-
Gene, inheritance, and pathogenesis: Ataxia Telangiectasia (AT) is an autosomal recessive ataxia. The disease is caused by mutation of ATM gene on chromosome 11q22.3. ATM gene is expressed in all tissues in the body. It plays a critical role in cell division of damaged cells. ATM genes phosphorylates P53, which facilitates apoptosis, and other tumor suppressor genes. ATM gene also regulates cell cycle progression from G1 to S and G2 to M, allowing the damaged cells to repair before the cell replicates. Therefore, those with an impaired ATM gene have difficulty with responding to DNA damage. ATM is also thought to have a possible role in mitochondrial hemostasis. Therefore, lack of ATM genes causes ataxia and other neurodegeneration symptoms. [74]
-
Clinical features
- Progressive neuronal degeneration
- Ataxia is usually the earliest sign and often occurs before the age of 4. Some children may be able to walk initially but then slowly regress. Unlike other ataxias, they tend to walk with a narrow base, and they prefer running over walking. Although their gait may be impaired, they do not have frequent falls. Once they are school age, the gross motor and fine motor skills begin to deteriorate and eventually become wheelchair bound [75].
- They also have other cerebellar symptoms including difficulty with eye coordination and dysarthria. They often develop oculomotor apraxia so they experience difficulty reading long sentences [76].
- Extrapyramidal signs can occur including choreoathetosis, dystonia, parkinsonism, and myoclonus.
- Progressive neuronal degeneration
Telangiectasia
-
Ocular telangiectasia is the hallmark this disease. It is commonly found in the eyes between the ages of 3 and 6 years old, but it can also be found in other parts of the body.
-
Abnormal pigmentation such as café au lait can be seen over the eyelids, face, ears, roof of the mouth, and other areas [77].
-
Immunodeficiency
- Immunoglobulin deficiencies especially immunoglobulin-A and immunoglobulin-G are common. They are subjected to recurrent respiratory infection, which can lead to interstitial lung disease [78]. Lymphopenia with reduced B lymphocytes number is also seen. Although they are immunocompromised, systemic bacteria, severe viral, or opportunistic infections are uncommon [108].
-
Malignancy
- Ten to 38% will develop at least one malignancy during their lifetime. Children are susceptible to blood related malignancies such as lymphoma or acute lymphoblastic leukemia. Prognosis is poor in these cases [109].
- Breast cancer especially in heterozygous population is common [110].
-
Cognitive impairments in AT present early [111].
-
Three quarter of children is found to have growth retardation. The cause is thought to be poor nutritional status and abnormal insulin -like growth factor 1 (IGF-1) secretion caused by pituitary abnormality. Some develop insulin resistance and hyperlipidemia.
-
Variant AT
- Patients with variant AT have milder cerebellar symptoms but have prominent extrapyramidal symptoms such as choreoathetosis, tremor, parkinsonism, dystonia, or myoclonus.
- Solid tumors are common, especially breast cancer. They also have ionizing radiation sensitivity.
- Ocular apraxia can be found but ocular telangiectasia may not be present.
- Immunodeficiency, infections, pulmonary issues, growth retardation, endocrine abnormality, or cognitive impairments are not present [112].
See the list below:
-
Diagnosis
- Clinical symptoms and ATM mutations on both alleles are confirmatory.
- Elevated alpha fetoprotein that is two standard deviation above the normal limit is a supportive of diagnosis [113].
- Radiation induced chromosomal breakage in cultured cells is supportive of diagnosis.
See the list below:
-
Treatment
- There is no disease modifying treatment or cure currently. The median age of death is 25 years old and the leading cause of death is progressive pulmonary disease due to recurrent respiratory infection or malignancy. A multidisciplinary approach and supportive therapy are essential.
- Riluzole and amantadine have been shown to improve ataxia in some hereditary ataxia, but there are no randomized placebo control studies for AT specifically. Focal dystonia can be treated with botulinum neurotoxin A while generalized dystonia can be treated with anticholinergic or GABA mimics such as baclofen and clonazepam. Intrathecal baclofen is not recommended. Evidence of tetrabenazine is limited. Some may have dopa responsive dystonia so a levodopa challenge can be attempted. Myoclonus can be treated with clonazepam, valproic acid, and levetiracetam. Tremor can be treated with propranolol and clonazepam. Efficacy of DBS for dystonia or tremor is not studied in this population.
- For infection, prevention is essential. Live vaccinations are not recommended although most children will have already received MMR by the time of diagnosis. Pneumococcal vaccination for children over the age of 3 years old is recommended. Prophylactic treatment with antibiotics may be warranted.
- Annual monitoring for total serum immunoglobulin G, M protein, and lymphocytes phenotyping is recommended. They have a low threshold for immunoglobulin replacement therapy.
- For malignancy, treat the underlying disease. Radiotherapy and radiomimic treatments such as bleomycin and neurotoxic agents should be avoided. Routine cancer screening is recommended. For heterozygous AT, breast cancer screening is recommended after the age of 25 and other malignancy screening prior to age of 50.
- Diabetes screening with A1C is recommended. Screening for lipids is not indicated as patients do not survive long enough to develop atherosclerotic disease.
Xeroderma pigmentosum
See the list below:
-
Gene, inheritance, and pathogenesis
This genetically heterogeneous disorder is due to a defect in DNA excision repair following UV exposure.
The condition differs from Cockayne syndrome because of the presence of skin tumors, absence of intracranial calcifications, and a different molecular defect. This disorder also has a poor prognosis.
-
Clinical features
Ataxia, chorea, and axonal polyneuropathy
Cutaneous photosensitivity and multiple cancers
Mental and motor retardation
Microcephaly
Sensorineural deafness
-
Laboratory findings: Defective DNA repair after ultraviolet radiation damage
Ataxia telangiectasia
This progressive, recessively inherited ataxia manifests in early childhood. It is more common in certain ethnic populations, including in those of Amish, Mennonite, Costa Rican, Polish, British, Italian, Turkish, Iranian, and Israeli descent.
-
Gene, inheritance, and pathogenesis: A defective truncated protein that belongs to the phosphatidylinositol-3 kinase family of proteins results from mutations that affect the ATM gene locus. This protein phosphorylates key substrates that are involved in DNA repair. The disease begins when patients are aged 1-3 years. No treatment is available other than supportive care and careful management of complications with modified chemotherapy
-
Clinical features
Choreoathetosis
Cutaneous and bulbar telangiectasia (present in teenagers and older individuals)
Immunodeficiency and increased susceptibility to infections
Oculomotor apraxia
Progressive ataxia and slurred speech
Susceptibility to cancer (eg, leukemia, lymphoma)
-
Laboratory findings
Molecular genetic testing is performed for mutations affecting the ATM gene locus (11q22.3). For those patients in whom mutations cannot be identified, other supportive laboratory evidence must be sought
Elevated (>10 ng/mL) serum alpha-fetoprotein is found in 90-95% of patients.
Findings on colony survival assay, ie, colony formation of a lymphoblastoid cell line following irradiation, are abnormal.
Karyotyping abnormalities involve 7-14 chromosomal translocation in 5-15% of cells after phytohemagglutinin stimulation of lymphocytes in peripheral blood.
Breakpoints result in translocation at the 14q11 and 14q32 sites.
Ataxia telangiectasia–like disorders
This group includes the following disorders: ataxia with oculomotor apraxia type 1 (AOA1), ataxia with oculomotor apraxia type 2 (AOA2), and ARSACS. [45]
-
Ataxia with oculomotor apraxia type I
Gene, inheritance, and pathogenesis: The disorder begins in childhood, proceeding to loss of ambulation in 7-10 years. The gene locus at 9p13.3 codes for a protein aprataxin. Mutations in this gene are pathogenic. The protein appears to have a role in DNA repair.
Clinical features
Progressive cerebellar ataxia
Oculomotor apraxia progressing to complete ophthalmoplegia
Motor neuropathy, progressive distal amyotrophy
Normal cognition in Portuguese families, decline in cognition noted in Japanese families
Laboratory findings
Hypoalbuminemia
No specific diagnostic tests available
-
Ataxia with oculomotor apraxia type 2
Gene, inheritance, and pathogenesis: The disorder begins in the second decade of life. The gene locus is 9q34, and the gene product is called senataxin. The protein is thought to function as a helicase involved in various aspects of DNA transcription and repair, RNA maturation, and termination.
Clinical features
Axonal sensorimotor neuropathy
Oculomotor apraxia is an inconsistent feature.
Laboratory findings
Cerebellar atrophy on imaging
Elevated alpha-fetoprotein
Table 6. Recessively Inherited Chronic/Progressive Ataxias Associated with DNA Repair Defects (Open Table in a new window)
Disorder/Syndrome |
Neurologic Phenotype |
Inheritance |
Gene Locus |
Gene Product/Biochemical Defect |
Cockayne syndrome type A |
Progressive ataxia plus Early onset severe syndrome |
Autosomal recessive |
5q11 |
ERCC8 |
Cockayne syndrome type B |
Progressive ataxia plus Classical type |
Autosomal dominant |
10q11-q21 |
ERCC6 |
Xeroderma pigmentosum |
Progressive ataxia plus |
Autosomal recessive |
Genetically heterogeneous with several complementation groups identified 9q34 locus (A) Other complementation groups involved are 2q21 (B & CS); 3p25.1 (C); 19q13.2(D); Unknown (E); 16p13 (F); 13q32-33 (G & CS) |
Mutations result in either defective damage-specific DNA-binding protein or defective excision repair (ERCC) Neurologic manifestations beginning in childhood relate to complementation group |
Ataxia Telangiectasia |
Progressive ataxia plus |
Autosomal recessive |
11q22-q23 |
ATM gene Product belongs to the P-13 kinase family of proteins involved in DNA damage recognition |
Ataxia with oculomotor apraxia type 1 (AOA1) |
FRDA-like hypoalbuminemia |
Autosomal recessive |
9p13.3 |
Aprataxin (APTX) Role in single-stranded DNA repair |
Ataxia with oculomotor apraxia type 2 (AOA2) Changed to autosomal recessive cerebellar ataxia (SCAR1) |
Ocular apraxia is an inconsistent feature. Ataxia Distal amyotrophy Peripheral neuropathy |
Autosomal recessive |
9q34 |
Senataxin (SETX) Protein involved in RNA maturation and termination |
Recessively inherited ataxias associated with protein translation/folding defects
Recessively inherited ataxias associated with protein translocation/folding defects are discussed below.
Spastic ataxia of Charlevoix-Saguenay
See the list below:
-
Gene, inheritance, and pathogenesis
ARSACS is an autosomal recessive spastic ataxia of Charlevoix-Saguenay region. This is an early-onset ataxia, manifesting in infancy or early childhood, with a high prevalence in the Charlevoix-Saguenay region of northeastern Quebec.
The estimated carrier frequency in Charlevoix-Saguenay region is 1/22. It has also been described in other regions of the world such as Mediterranean areas and Japan. Mutations in the SACSIN gene encode a protein sacsin that is believed to function as a chaperone involved in protein folding.
-
Clinical features
Progressive ataxia with pyramidal, cerebellar, and distal neuropathy sensorimotor neuropathy
Nystagmus
Slurred speech
Hypermyelinated retinal nerve fibers leading to retinal striations
Skeletal abnormalities, including swan neck–like deformities of the fingers, pes cavus, and hammer toes
-
Laboratory findings
Decreased sensory nerve conduction velocities (NCV)
Decreased motor NCV
Loss of large myelinated fibers on nerve biopsy
Leukoencephalopathy with vanishing white matter (van der Knaap syndrome)
See the list below:
-
Gene, inheritance, and pathogenesis
Leukoencephalopathy with vanishing white matter (VWM) has an autosomal recessive inheritance with an age-dependent penetrance.
The gene is located on band 3q27. [46] The mutation involves a gene that codes for the eukaryotic translation initiation factor (eIF2B). The gene likely controls regulation of translation under conditions of stress. No effective treatment is known to halt progression of the disorder, although symptomatic and supportive measures can improve the quality of life. [47, 48]
-
Clinical features
Cerebellar ataxia and spasticity are prominent.
Chronic progressive neurologic deterioration and episodic exacerbation follow in late infancy or early childhood.
Episodes of deterioration follow minor infection and head trauma, leading to periods of lethargy or coma.
Cognitive ability may show decline but is relatively preserved compared to the severity of motor deficit.
Initial motor and mental development is normal or mildly delayed.
Optic atrophy and epilepsy may be additional features.
-
Laboratory findings
Cerebellar atrophy varies from mild to severe and primarily involves the vermis.
Elevated CSF glycine is a marker for this disorder.
MRI indicates symmetric involvement of the cerebral hemispheric white matter, which acquires a signal intensity close to or the same as CSF on proton density, T2-weighted, T1-weighted, and fluid-attenuated inversion recovery images.
Magnetic resonance spectroscopy shows a significant decrease to near absence of normal signals from the white matter, except for lactate and glucose (the signals of which become more prominent with disappearance of other normal signals). Signals over the cortex remain relatively normal.
Pathologic studies confirm white matter rarefaction and loss of myelinated white fibers. Microcystic changes are reported in the periventricular white matter.
4H syndrome
See the list below:
-
4H syndrome is a recessively inherited phenotype with distinctive clinical features and a hypomyelinating leukodystrophy. To date, no gene locus or mutations have been identified.
-
Clinical features
Early onset progressive ataxia
Short stature
Hypodontia
Delayed puberty secondary to gonadal dysfunction
-
Laboratory
MRI shows white matter signal abnormalities consistent with central hypomyelination and cerebellar atrophy.
Sural nerve biopsy shows debris-lined myelin clefts, vacuolar disruption, and loss of normal myelin periodicity.
Table 7. Recessively Inherited Chronic/Progressive Ataxias Associated with Protein Translation and Folding Defects (Open Table in a new window)
Disorder/Syndrome |
Neurologic Phenotype |
Inheritance |
Gene Locus |
Gene Product/Biochemical Defect |
Autosomal recessive spastic ataxia of Charlevoix-Saguenay |
Chronic ataxia Spasticity Retinal abnormalities |
Autosomal recessive |
13q11 |
SACS gene codes for sacsin, which is involved in chaperone-mediated protein folding |
Leukoencephalopathy with VWM |
Progressive ataxia Spasticity Optic atrophy Seizures |
Autosomal recessive |
3q27 |
Mutations affect eIF2B |
4H syndrome |
Short stature Slowly progressive ataxia Hypogonadism Hypomyelination hypodontia |
Autosomal recessive |
Not known |
Not known |
Recessively inherited chronic/progressive ataxias associated with inherited enzymatic defects
Recessively inherited chronic/progressive ataxias associated with inherited enzymatic defects are discussed below.
Refsum disease
See the list below:
-
Gene, inheritance, and pathogenesis: This autosomal recessive disorder is associated with impaired oxidation of phytanic acid. Elevated phytanic acid levels in the nervous system are associated with neurotoxicity.
-
Clinical features
Onset in the second to third decade of life
Cerebellar ataxia (may be superimposed in some patients)
Early presentation of night blindness and pigmentary degeneration of the retina
Polyneuropathy with elevated CSF protein
Sensorineural deafness
Skin (ichthyosis) and cardiac (arrhythmia) abnormalities
-
Laboratory findings
Cultured fibroblasts show reduced ability to oxidize phytanic acid.
Elevated phytanic acid levels in the plasma and urine are diagnostic.
-
Treatment: Refsum disease has a relapsing-remitting course. Drastic reduction in dietary phytanic acid (supplemented by plasmapheresis) at onset can ameliorate the neuropathy and possibly other clinical abnormalities.
Cerebrotendinous xanthomatosis
See the list below:
-
Gene, inheritance, and pathogenesis: This autosomal recessive disorder is caused by a defect in bile acid synthesis. Cholestanol accumulates in the tissues, including the nervous system. The defect is due to deficiency of hepatic sterol 27-hydroxylase, a mitochondrial enzyme.
-
Clinical features
Palatal myoclonus and seizures
Peripheral neuropathy
Progressive ataxia with mental decline
Pseudobulbar palsy
Tendon xanthomas
Cataracts
-
Laboratory findings
Elevated cholestanol and Apo-B in CSF
Low plasma cholesterol; elevated plasma cholestanol
Low-to-absent chenodeoxycholic acid in the bile
-
Treatment: Lifelong oral administration of chenodeoxycholic acid (750 mg/d) is effective if initiated early (see chenodiol). A 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor also can be added to inhibit cholesterol biosynthesis.
Biotinidase deficiency
See the list below:
-
Gene, inheritance, and pathogenesis: Because of the lack of free biotin, biotinidase deficiency results in dysfunction of 3 mitochondrial carboxylases. It is recessively inherited, and the underlying defect involves mutations of the 3p25 locus for biotinidase.
-
Clinical features
Delayed presentation (second year of life)
Intermittent ataxia, sensorineural hearing loss
Myoclonic seizures, developmental delay
Skin rashes, alopecia
-
Laboratory findings
Organic aciduria (eg, elevated beta-hydroxyisovalerate, lactate, beta-methylcrotonylglycine, beta-hydroxypropionate, methylcitrate)
Mild hyperammonemia
Diffuse cerebral and cerebellar atrophy on cranial MRI
Metabolic acidosis, lactic acidosis
Biotinidase activity in serum and fibroblasts
Mutation analysis
-
Treatment
Biotin 5-20 mg/d PO is remarkably effective in reversing neurologic and cutaneous symptoms.
Hearing and visual dysfunction may be resistant to treatment.
L-2-hydroxyglutaricaciduria
See the list below:
-
Gene, inheritance, and pathogenesis: This autosomal recessive inherited defect is characterized by excessive excretion of L-2-hydroxyglutaric acid in the urine. The precise molecular basis is not well established. The clinical course is of slowly progressive neurodegenerative disorder.
-
Clinical features
Age of onset from 6-20 years
Presence of cognitive delay and epileptic seizures
Progressive ataxia, dysarthria, and extrapyramidal dysfunction
Added features of short stature and macrocrania
-
Laboratory findings
Elevated 2-hydroxyglutaric acid in plasma, urine, and CSF
Elevated lysine in plasma and CSF
Highly specific MRI pattern showing subcortical leukoencephalopathy with bilateral high signal intensity in dentate nuclei and putaminal regions
Succinic-semialdehyde dehydrogenase deficiency
See the list below:
-
Gene, inheritance, and pathogenesis: Succinic-semialdehyde dehydrogenase deficiency (SSADH) is a recessively inherited disorder affecting the gamma-aminobutyric acid (GABA) degradation pathway. Although it is characterized by excretion of large amounts of 4-hydroxybutyric acid in the urine, phenotype varies widely. [49]
-
Clinical features
Ataxia
Hypotonia
Nonspecific neurologic features such as cerebral palsy and developmental delay
Psychomotor retardation, language delay
-
Laboratory findings
Elevated 4-hydroxybutyric acid in plasma, urine, and CSF
High free GABA in CSF
Cerebellar atrophy on MRI
-
Treatment
L-carnitine supplementation has been tried with improvement in muscle tone.
Vigabatrin, an inhibitor of GABA transaminase, has proven effective in low doses of 25 mg/kg/d.
Late-onset sphingolipidoses
These complex biochemical defects are related to specific deficiencies of lysosomal enzymes (see Table 8 below). The brain and other tissues such as the liver store abnormal sphingolipids. The presentation is a combination of cognitive deterioration, seizures, and gait abnormalities due to a combination of pyramidal features (spasticity), cerebellar dysfunction (ataxia), extrapyramidal features (eg, dystonia), choreoathetosis, and ophthalmologic abnormalities. Ataxia almost never is the sole clinical symptom. Other systemic features can include coarse facies, organomegaly, and dysostosis multiplex. Because these disorders are progressive, symptoms and signs can be seen in combination. The disorders are autosomal recessive. Skin fibroblast examination under electron microscope is an effective screening tool. Definitive diagnosis can be established by lysosomal enzyme assay in leukocytes or cultured skin fibroblasts.
Congenital disorders of glycosylation
The congenital disorders of glycosylation (CDG) represent a new class of disorders that result from abnormalities of carbohydrate-deficient glycoproteins, particularly transferrin. The disorder has been reported from Scandinavian countries as well as other European countries. Most are autosomal recessive conditions; several (nearly 20 at the latest count) clinical and biochemical types have been characterized. Because glycoproteins are important constituents of the developing brain, CNS involvement and multisystem manifestations are frequent.
-
Gene, inheritance, and pathogenesis: CDG type 1a is caused by mutations affecting the enzyme phosphomannomutase; the gene locus is located on sub band 16p13.3. The enzyme is involved in the N-glycosylation pathway. Several other disorders involving the O-glycosylation pathway have now been recognized; the Walker-Warburg syndrome and the muscle-eye-brain disease are examples. For the purposes of the present discussion on ataxia the authors restrict discussion to CDG type 1a. The mortality rate is approximately 20% in the first 2 years. Only supportive treatment is available.
-
Clinical features
Stage of ataxia; mental deficiency during infantile and childhood stage
Delayed development, failure to thrive, hypotonia, and multisystem organ failure
Dysmorphic facial features, including prominent ears and nose
Fat pads over buttocks, abnormal patches of skin over thighs (orange peel skin), and inverted nipples (considered characteristic clinical features)
In the teenage years, evident lower limb atrophy and peripheral neuropathy
Severe intellectual disability and hypogonadism recognized in later years
-
Laboratory findings
Decreased serum glycoproteins
MRI showing striking pontocerebellar atrophy
Reduced thyroxine-binding globulin levels
Sialic acid, galactose, and N -acetylglucosamine deficiency in total serum glycoproteins
Synthesized proteins with fewer attached carbohydrate moieties than normal glycoproteins
Separation of proteins based on charge when an electric field is applied to serum
Sialotransferrins, a specific class of glycoproteins, behave differently in serum from patients with CDG than in serum from individuals without CDG; patients with CDG have less sialic acid, a negatively charged sugar.
The pattern of separation during electrophoresis (transferrin isoimmunoelectrophoresis) is considered diagnostic for this disorder.
Phosphomannomutase deficiency in leukocytes, fibroblasts, or liver
Consideration of molecular analysis of phosphomannomutase 2 gene (PMM2) in some subtypes
Marinesco-Sjögren syndrome
See the list below:
-
Gene, inheritance, and pathogenesis: Marinesco-Sjögren syndrome (MSS) is an autosomal recessive disorder. MSS is mapped to chromosome arm 5q31, but genetic heterogeneity is evident. In some families, mutations have been identified in the gene SIL1, [50] which encodes a nucleotide exchange factor for the heat-shock protein 70 (HSP70) chaperone HSPA5. The disorder is now thought to be a consequence of dysfunction of the endoplasmic reticulum and disturbed SIL1-HSPA5 interaction and protein folding. This disorder has overlapping features with lysosomal disorders. Ophthalmologic, skeletal, and gonadal abnormalities are frequently seen.
-
Clinical features
Microcephaly
Cataracts
Cerebellar ataxia
Mild-to-moderate intellectual disability
Neuromuscular weakness
Short stature
Hypergonadotropic hypogonadism
Skeletal anomalies of kyphosis, scoliosis, and coxa valga
Table 8. Recessively Inherited Chronic/Progressive Ataxias Associated with Inherited Enzyme Defects (Open Table in a new window)
| Disorder/Syndrome | Neurologic Phenotype | Inheritance | Locus/Gene |
|---|---|---|---|
| Refsum disease | Some have ataxia Retinitis pigmentosa Peripheral neuropathy Cardiac dysfunction Deafness |
Autosomal recessive | 10p13 PHYH |
| Cerebrotendinous xanthomatosis | Cerebellar ataxia Spinal cord involvement Pseudobulbar palsy Cataracts Palatal myoclonus Seizures Mental decline Tendon xanthomas |
Autosomal recessive | 2q35 CYP27A1 |
| Biotinidase deficiency | Intermittent ataxia Sensorineural hearing loss Seizures Developmental delay Skin rashes, alopecia Hearing loss Optic atrophy |
Autosomal recessive | 3q25.1 BTD |
| L-2 hydroxyglutaric acidemia | Intellectual disability Seizures Progressive ataxia and other cerebellar signs Macrocephaly |
Autosomal recessive | 14q21.3 L2HGDH |
| Succinic-semialdehyde dehydrogenase deficiency | Ataxia Seizures Hypotonia Developmental delay of motor, mental and language skills |
Autosomal recessive | 6p22.3 ALDH5A1 |
Late infantile and juvenile sphingolipidoses 1. Metachromatic leukodystrophy 2. Krabbe disease 3. Gaucher type III 4. Niemann-Pick C disease 5. GM2 gangliosidosis |
Progressive ataxia plus Seizures Psychomotor regression Spasticity Extrapyramidal features Supranuclear gaze palsies |
Autosomal recessive | 1. 22q13.3 -ARSA 2. 14q31.3- GALC 3. 1q22- GBA 4. 18q11.2– NPC1 5. 5q33.1- GM2A |
| Congenital disorders of glycosylation type Ia | Psychomotor retardation Axial hypotonia Abnormal eye movement Peripheral neuropathy Cerebellar ataxia |
Autosomal recessive | 16p13.2 PMM2 |
| Marinesco-Sjögren syndrome | Ataxia Congenital cataract Hypotonia Myopathy Delay of psychomotor development |
Autosomal recessive | 5q31.2 SIL1 |
Recessively inherited ataxias associated with mitochondrial cytopathies
Recessively inherited ataxias associated with mitochondrial cytopathies are discussed below.
Neuropathy, ataxia, retinitis pigmentosa, and peripheral neuropathy syndrome (maternal inheritance)
Gene, inheritance, and pathogenesis: Neuropathy, ataxia, retinitis pigmentosa, and peripheral neuropathy (NARP) syndrome is a mitochondrial disorder that displays maternal inheritance. Affected individuals present with features of cerebellar ataxia, seizures, cognitive impairment, and peripheral neuropathy. The condition carries a variable phenotype and also may occur sporadically. The underlying defect involves a mitochondrial adenosine triphosphate (ATP) synthase gene (subunit 6) affecting nucleotide 8993, mutations of which also can result in the Leigh syndrome phenotype. The diagnosis can be confirmed by mitochondrial DNA mutation analysis.
Leigh disease
See the list below:
-
Gene, inheritance, and pathogenesis: This disorder has distinct neuropathologic findings, highly variable clinical presentation, and can be caused by multiple biochemical and molecular genetic defects. Autosomal recessive inheritance and maternal inheritance (mutations in mitochondrial DNA) patterns exist.
-
Clinical features: Clinical features include protean manifestations due to multifocal lesions in the brainstem, thalamus, and cerebellum; the most important of these are as follows:
Oculomotor - Nuclear or supranuclear ophthalmoplegia, central nystagmus with rotary and horizontal components
Course - Relapsing-remitting course, rarely progressively fatal
Respiratory - Characterized by unexplained hyperventilation, apnea, and irregular respiration (air hunger)
Neurologic - Truncal ataxia, incoordination, and intention tremor evident as child begins to walk
-
Laboratory findings
Characteristic symmetric lesions can be demonstrated in the thalamus, putamen, and globus pallidus on T2-weighted MRI sequences. The lesions also are distributed in the brainstem and cerebellum.
Lactate and pyruvate are elevated in the CSF.
Perform enzyme function assays on cultured fibroblasts, muscle, or liver tissue. Frequently, more than one of these tissues should be assayed because of the lack of correlation between enzyme activities in muscle and skin.
Hyperammonemia, hypoglycemia, and organic aciduria are not present.
Multiple mitochondrial enzymes have been demonstrated to be affected in this disorder, particularly the pyruvate dehydrogenase (PDH) complex, cytochrome c oxidase, and the mitochondrial adenosine triphosphatase (ATPase) 6 gene.
Neuropathologic lesions show incomplete necrosis and spongiform changes in the neuropil with relative preservation of the neurons, resulting in a spongiosis. Vascular proliferation also occurs, and white matter changes can be seen.
-
Treatment: No treatment is known to actually benefit patients. Vitamin B1 (thiamine) supplementation has been administered without documented benefit. Recently, the ketogenic diet has been reported to be useful in treating patients with PDH complex deficiency.
Coenzyme-Q10–associated ataxia
CoQ-10 is involved in facilitation of electron transfer between the various dehydrogenases and cytochromes participating in the respiratory chain and oxidative phosphorylation reaction. Ubiquinone deficiency presents with many different clinical phenotypes ranging from myopathy to Leigh's disease.
-
Gene inheritance and pathogenesis: Autosomal recessive, genetic heterogeneity is likely. Mutations (missense) in the CABC1 gene, also called COQ8 or ADCK3, coding for a putative protein kinase in the ubiquinone biosynthesis pathway have recently been shown to be associated with this form of CoQ-10 deficiency. [51]
-
Clinical features
More than 20 patients have been described with a recessively inherited form of muscle CoQ-10 deficiency who present with a slowly progressive ataxia in childhood, associated with cerebellar atrophy. [52]
Associated features include developmental delay, intellectual disability, and seizures.
-
Laboratory
A few patients demonstrate elevations in plasma lactate.
Decreased CoQ concentration in muscle or fibroblasts.
Table 9. Recessively Inherited Chronic/Progressive Ataxias Associated with Mitochondrial Cytopathies (Open Table in a new window)
Disorder/Syndrome |
Neurologic Phenotype |
Inheritance |
Gene Product/Biochemical Defect |
NARP syndrome |
Progressive ataxia plus |
Maternal inheritance |
Mitochondrial ATP 6 NARP 8993 mutation causing base substitution T-G or T-C at nucleotide position 8993 |
Leigh disease |
Progressive ataxia plus Lactic acidosis |
Autosomal recessive/maternal inheritance |
Multiple biochemical and molecular defects underlie the condition, eg, PDHC deficiency, cytochrome oxidase C deficiency, mitochondrial ATPase 6 |
CoQ-10 responsive ataxia |
Progressive ataxia in childhood Developmental delay Seizures Cerebellar atrophy on MRI |
Autosomal recessive |
Mutations in the gene CABC1 or ADCK3 are described. The gene codes for a putative protein kinase associated with ubiquinone biosynthesis. |
Progressive Ataxias With Polymyoclonus and Epileptic Seizures
Progressive myoclonic epilepsies
The progressive myoclonic epilepsies (PMEs) constitute a group of seizure disorders with phenotypic features of myoclonic and other generalized seizures, ataxia, and cognitive defects. These features occur in variable combinations that progress over time. These disorders are often difficult to distinguish on purely clinical grounds.
Dodecamer repeat expansions
Dodecamer repeat expansions are discussed below.
Myoclonic epilepsy of Unverricht and Lundborg (EMP1)
See the list below:
-
Gene, inheritance, and pathogenesis: PME of the Unverricht-Lundborg type (EPM1) is an autosomal recessive disease caused by mutation in the cystatin B gene (CSTB). The protein cystatin B is a cysteine protease inhibitor. The mutation results from an unstable dodecamer repeat expansion in the promoter region of the CSTB gene.
-
Clinical features
Onset is usually around 10 years of age
Ataxia
Progressive myoclonus, generalized tonic-clonic seizures
Mild mental deterioration
Stimulus and photo-sensitive myoclonus
Disease usually stabilizes in early adulthood, and in some cases myoclonus and ataxia may even improve
-
Diagnosis
EEG shows light-sensitive, generally synchronous, spike and wave
Giant somatosensory evoked potentials can be elicited
-
Treatment
N-acetylcysteine has been shown to reduce seizures, but not myoclonus or ataxia; however, the response is variable
Phenytoin aggravates symptoms
Levetiracetam has been useful in the treatment of myoclonus [97, 98, 99, 100]
Inherited enzyme defects
Inherited enzyme defects are discussed below.
Lafora body disease
See the list below:
-
Gene, inheritance, and pathogenesis: Lafora disease is also known as progressive myoclonic epilepsy-2 (EPM2A). It is an autosomal recessive disorder. EPM2 is linked to 6q22 and the malin gene (NHLRC1)or the laforin gene (EPM2A).
-
Clinical features
Onset usually in adolescence
Initial symptoms can be vague such as headache, school performance difficulties, myoclonic jerks. Some symptoms are more obvious such as visual hallucinations and seizures
There can be many seizures types including generalized tonic-clonic seizures, simple partial occipital seizures, partial seizures, absence seizures and myoclonic seizures
Patients have decline in mental status
Myoclonic jerks
Progressive worsening myoclonic and occipital seizures with visual signs
Disease is usually fatal
-
Diagnosis
Axillary skin biopsy shows periodic acid-Schiff (PAS)–positive inclusion bodies are found in the apocrine glands. These findings are considered diagnostic [101]
Neuronal ceroid lipofuscinosis-2 (CLN2)
See the list below:
-
Gene, inheritance, and pathogenesis: Neuronal ceroid lipofuscinosis (NCL/CLN) describes autosomal recessive disorders in which characteristic storage material is identified within neurons, resulting in their degeneration. NCLs are a group of progressive neurodegenerative disorders that share several clinical features, particularly the presence of seizures and progressive dementia. Several genetically distinct subgroups have been determined based on age at presentation. Each subgroup has a characteristic ultrastructural appearance of the intracellular lipopigment. The gene for the classic late infantile-onset form (TPP1) maps to band 11p15.
-
Clinical features
Onset in young childhood (2–4 years of age)
Ataxia
Dementia
Myoclonic seizures, atypical absence seizures, GTC seizures, other seizure types
Visual impairment
Mental regression
Myoclonus
-
Diagnosis
CT scanning and MRI show predominantly cerebellar atrophy. See image below.
 Magnetic resonance imaging study of the brain in a patient with neuronal ceroid lipofuscinosis showing cerebellar atrophy on sagittal view.
Magnetic resonance imaging study of the brain in a patient with neuronal ceroid lipofuscinosis showing cerebellar atrophy on sagittal view.
Electron microscopic examination of skin or other tissue shows typical curvilinear inclusions
Patients have enlarged visual evoked potential
Photic stimulation on EEG produces high-amplitude spikes

Mitochondrial cytopathies
Mitochondrial cytopathies are discussed below.
Myoclonic epilepsy with ragged red fibers (MERRF)
See the list below:
-
Gene, inheritance, and pathogenesis: MERRF is a disorder that affects different parts of the body, especially the nervous system and muscles. Multiple mitochondrial gene mutations have been found. Some examples of genes affected include MTTK, MTTL1, MTTH, MTTS1, MTTS2, and MTTF. It has maternal inheritance, but phenotype can vary widely. The mutations cause deficient mitochondrial energy production. An A-to-G transition mutation at nucleotide pair 8344 in human mitochondrial DNA has been identified in most patients. The mutation creates a specific restriction site on the tRNALys gene, producing defects in complex I and IV enzymes of the oxidative phosphorylation system. Myriad cell functions are involved in the control of excitability and are energy dependent. Thus, deficient energy production or utilization can lead to neurologic dysfunction in a variety of ways.
-
Clinical features
Myoclonic seizures
Other seizure types
Cerebellar ataxia
Myopathy
Sensorineural deafness
Short stature
Can also have optic atrophy, peripheral neuropathy, spasticity, and dementia
-
Diagnosis
CT scan shows cerebral atrophy and bilateral basal ganglia calcification
Muscle biopsy shows ragged red fibers, which are caused by the subsarcolemmal aggregation of mitochondria
Lactate levels are elevated in blood and cerebrospinal fluid
-
Treatment
The seizure disorder can be treated with conventional anticonvulsant therapy [104]
Table 10. Progressive Ataxias with Myoclonus and Epileptic Seizures (Open Table in a new window)
Type |
Unverricht-Lundborg syndrome |
Neurologic Phenotype |
Inheritance |
Locus |
Dodecamer repeat expansion |
Unverricht-Lundborg syndrome |
Myoclonus Ataxia Seizures |
Autosomal recessive 21q22.2 |
21q22.2 CSTB |
Inherited enzyme defect |
Lafora body disease |
Myoclonus Ataxia Seizures Mental decline |
Autosomal recessive 2A- 6q24.3 EPM2A |
2B – 6p22.3 NHLRC1 |
Inherited enzyme defect |
Neuronal ceroid lipofuscinosis |
Myoclonus Ataxia Seizures Visual impairment |
Autosomal recessive |
11p15.4 TPP1 |
Mitochondrial cytopathy |
MERRF |
Myoclonic epilepsy Ataxia Hearing disturbance Mental deterioration Muscle atrophy |
Maternal inheritance |
Multiple mutations, including: MTTK, MTTL1, MTTH, MTTS1, MTTS2, MTTF, MTND5 |
Other Disorders
Angelman syndrome
See the list below:
-
Gene, inheritance, and pathogenesis: This is a disorder caused by abnormalities of imprinting. It manifests with significant gait abnormalities and ataxia with characteristic pattern, previously known as happy puppet syndrome (this term is no longer used). A number of different pathogenetic mechanisms such as loss of maternal allele, paternal uniparental disomy, mutation of the ubiquitin protein ligase E3A gene ( UBE3A) account for different subsets of Angelman syndrome.
-
Clinical features
Happy disposition with paroxysmal laughter
Severe intellectual disability, severe limitation in speech and language
Wide-based gait, ataxia
Some patients have microcephaly
Hypopigmentation or fair skin, reduced retinal pigmentation (usually blue eyes)
Significant seizures
Prognathia
Characteristic arm position with wrist and elbow flexion
-
Diagnosis
DNA methylation abnormalities in the 15q11-13 region
Deletions in 15q11-13 region using fluorescent in situ hybridization (FISH)
Uniparental disomy studies
UBE3A mutation on DNA analysis
Mild cortical atrophy on CT scanning or MRI
EEG abnormalities that are considered as highly characteristic [105]
Fragile X syndrome/ataxia (FXTAS)
See the list below:
-
Gene, inheritance, and pathogenesis: FXTAS is a disorder caused by a trinucleotide repeat in FMR1 gene. Carriers of premutation alleles (55-200 CGG repeats) of fragile X mental retardation 1 (FMR1) are now being identified with one (or more) distinct clinical disorders, including mild cognitive delay and/or behavioral deficits on the fragile X spectrum and a neurodegenerative disorder among older adult carriers. This is known as the fragile X–associated tremor/ataxia syndrome (FXTAS). Awareness of these clinical presentations is important for physicians involved in the care of patients with fragile X syndrome but also more broadly for neurologists caring for adults with tremor, gait ataxia, and parkinsonism. Female carriers of the FMR1 premutation who present with symptoms of tremor and ataxia do not develop features of dementia, unlike males with FXTAS. This protective effect in female carriers remains unexplained at present.
-
In the fragile X tremor ataxia syndrome, up to 40% of males and 4-8% of females carrying the premutation can be symptomatic. In the premutation states, the degree of FMR protein (FMRP) amounts are not decreased. The levels may be near normal; however, the amount of mRNA expression is noted to be 8-10 times elevated. This is not seen with the full mutation. The tremor ataxia syndrome is thought to be related to mRNA accumulation and resultant toxicity. [106, 107]
Approach to Patients With a Suspected Inherited Ataxia
The assessment of such a patient involves obtaining a detailed clinical history complemented by an appropriate neurologic examination that delineates the following information:
-
Age of onset
-
Mode of onset (ie, acute, subacute, chronic)
-
Sex
-
Natural history (ie, nonprogressive/static, episodic, progressive)
-
Associated symptoms/signs that provide localizing information
Presence of dystonia or chorea suggesting involvement of the striatum
Proprioceptive dysfunction suggesting involvement of spinocerebellar pathways
Visual deficits (retinitis pigmentosa), auditory involvement (Refsum disease)
Cognitive dysfunction possible early and/or late
-
Other systemic features
Dysmorphic features and associated congenital malformation may suggest a specific association or clinical syndrome.
Cardiac (Friedreich ataxia), renal (NPCA), and cutaneous (xeroderma pigmentosa) features are examples.
-
Family history and pedigree analysis provides diagnostic clues and information on possible patterns of inheritance, which are useful for planning investigations and genetic counseling.
Once a specific clinical phenotype is delineated, the investigative process can be initiated based on the clinical features. The initial step involves obtaining specific neuroimaging studies; MRI is often preferable because it can provide detailed information helpful in anatomic localization (ie, signal changes in the cortex, white matter, cerebellum, striatum, and brainstem), and patterns of involvement in some conditions can be diagnostic. In mitochondrial cytopathies, magnetic resonance (MR) spectroscopy (ProtonMRS) can demonstrate an elevated lactate peak and can complement the findings on MRI. A karyotype (demonstrating deletions, duplications, and chromosomal rearrangements), specialized cytogenetic studies (as in Angelman syndrome), and DNA-based molecular diagnostics (as in SCAs, fragile X syndrome, and Angelman syndrome) can be utilized to provide rapid turnaround times for diagnosis.
Metabolic screening involves tests such as quantitative studies for plasma lactate, ammonia, carnitine levels, amino acids in blood and urine, urine analysis for organic acid and acylglycines (stable isotope dilution gas chromatography–mass spectrometry [GC/MS]), plasma acylcarnitines (tandem mass spectrometry [MS/MS]), and assays for sialotransferrins (isoelectric focusing of serum transferrins) should be used selectively after consultation with a metabolic geneticist. A schematic approach is suggested (see image below).
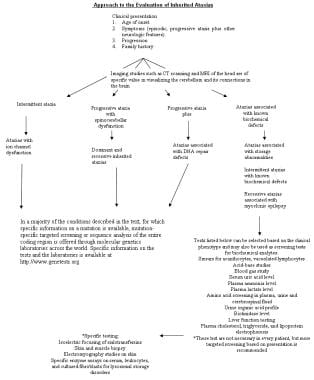 Approach to the biochemical evaluation of inherited ataxia. Screening tests should be targeted to clinical presentations.
Approach to the biochemical evaluation of inherited ataxia. Screening tests should be targeted to clinical presentations.
Conclusions
With the completion of the Human Genome Project, newer gene discoveries have ushered in an era where making diagnoses is not limited to clinical aspects but also relies on establishing a molecular basis. The identification of gene-protein links to specific cellular pathways adds to the understanding and eventually guide the way for future therapeutic advances. When assessing a child or adult with ataxia, the differential diagnosis always must include biochemical defects. The age of onset, mode of presentation, family history, and presence or absence of other neurologic signs are involved heavily in determining the screening and specific tests used in the evaluation (see image below).
Approach to the biochemical evaluation of inherited ataxia. Screening tests should be targeted to clinical presentations.
Many of these conditions are progressive and neurodegenerative, with no treatment currently available. Identification of specific defects, such as ataxia with selective vitamin deficiency, provides treatment options for disorders that are eminently treatable. For other incurable disorders, such as Friedreich ataxia, treatment approaches such as antioxidant therapy may prolong life and lead to reduction in morbidity. Idebenone, a synthetic analogue of coenzyme Q has been tried with beneficial effects on ADL scores on both cardiac hypertrophy as well as neurologic symptoms in preliminary trials. Supportive therapy, management of associated complications, and the role of support groups cannot be overemphasized for these families who have to deal with truly challenging medical needs.
Support groups
International Network of Ataxia Friends (INTERNAF)
2600 Fernbrook Lane; Suite 119
Minneapolis, MN 55447
Phone: 763-553-0020
Fax: 763-553-0167
Email: naf@ataxia.org
Spinocerebellar Ataxia: Making an Informed Choice about Genetic Testing
Euro-ataxia (European Federation of Hereditary Ataxias)
Boherboy, Dunlavin
Co Wicklow, Ireland
Phone: +353 45 401218
Fax: +353 45 401371
Email: mary.kearneyl@euro-ataxia.org
NCBI Genes and Disease - Spinocerebellar ataxia
WE MOVE (Worldwide Education and Awareness for Movement Disorders)
204 West 84th Street
New York, NY 10024
Phone: 800-437-MOV2 (800-437-6683)
Fax: 212-875-8389
Email: wemove@wemove.org
-
Putative pathogenic mechanisms of cerebellar disease.
-
Approach to the biochemical evaluation of inherited ataxia. Screening tests should be targeted to clinical presentations.
-
Magnetic resonance imaging study of the brain in a patient with neuronal ceroid lipofuscinosis showing cerebellar atrophy on sagittal view.
Tables
- Table 1. Nonprogressive Congenital Ataxias
- Table 2. Episodic Ataxias
- Table 3. Intermittent Ataxias Related to Enzyme Defects
- Table 4. Dominantly Inherited Chronic/Progressive Ataxias
- Table 5. Recessively Inherited Chronic/Progressive Ataxias with Spinocerebellar Dysfunction
- Table 6. Recessively Inherited Chronic/Progressive Ataxias Associated with DNA Repair Defects
- Table 7. Recessively Inherited Chronic/Progressive Ataxias Associated with Protein Translation and Folding Defects
- Table 8. Recessively Inherited Chronic/Progressive Ataxias Associated with Inherited Enzyme Defects
- Table 9. Recessively Inherited Chronic/Progressive Ataxias Associated with Mitochondrial Cytopathies
- Table 10. Progressive Ataxias with Myoclonus and Epileptic Seizures
Disorder/Syndrome |
Phenotype* |
Inheritance |
NPCA with or without cerebellar hypoplasia |
Early hypotonia Delayed motor and speech development |
Autosomal recessive
Autosomal dominant
X-linked recessive
Sporadic |
NPCA with posterior fossa malformations (eg, Dandy Walker syndrome) |
Variable association with hydrocephalus Delays in motor development Cognitive delay |
N/A |
Ataxia syndromes, multiple congenital anomalies, and cerebellar hypoplasia (eg, Joubert syndrome, Varadi syndrome, COACH syndrome) |
Encephalo-oculo-hepato-renal anomalies with recognized association patterns of anomalies |
Autosomal recessive
Autosomal dominant
X-linked |
Ataxia syndromes with cerebellar hypoplasia (eg, Gillespie syndrome) |
Partial aniridia Hypogonadotrophic hypogonadism External exophthalmoplegia |
Autosomal recessive |
*Gait ataxia is a constant feature. |
||
Disorder/Syndrome |
Phenotype* |
Inheritance |
Locus/Gene |
EA1 |
Intermittent ataxia |
Autosomal dominant |
12q13 KCNA1 |
EA2 |
Intermittent ataxia |
Autosomal dominant |
19q13 CACNA1A |
EA3 |
Intermittent ataxia with vertigo and tinnitus |
Autosomal dominant |
1q42 |
| EA4 | Intermittent ataxia, vertigo, diplopia | Autosomal dominant | Unknown |
| EA5 | Intermittent vertigo and ataxia lasting several hours | Autosomal dominant | 2q23.3 CACNB4 |
| EA6 | Intermittent ataxia, seizures, migraine and alternating hemiplegia |
Autosomal dominant | 5p13.2 SLC1A3 |
| EA7 | Vertigo, weakness, dysarthria | Autosomal dominant | 19q13 |
Disorder/Syndrome |
Phenotype* |
Inheritance |
Locus/Gene |
Maple syrup urine disease |
Intermittent ataxia |
Autosomal recessive |
1p21.2 – DBT 6q14.1 – BCKDHB 19q13.2 – BCKDHA |
Hartnup disease |
Intermittent ataxia |
Autosomal recessive |
5p15.33 SLC6A19 |
Pyruvate dehydrogenase deficiency |
Intermittent ataxia Lactic acidosis |
X-linked recessive |
Xp22.12 |
Pyruvate carboxylase deficiency |
Intermittent ataxia Lactic acidosis |
Autosomal recessive |
11q13.2 PC |
Defects of mitochondrial fatty acid beta-oxidation |
Intermittent ataxia Metabolic acidosis Elevated ammonia |
Autosomal recessive |
N/A |
Late-onset urea cycle defects Argininosuccinic acidemia Carbamyl phosphate synthetase deficiency Citrullinemia Ornithine transcarbamoylase deficiency Argininemia |
Intermittent ataxia Episodic encephalopathy |
Autosomal recessive |
7q11.21 (arginosuccinate lyase) 2q34 (carbamoyl-phosphate synthetase I) 9q34.11 (arginosuccinate synthetase) Xp11.4 (ornithine carbamoyltransferase) 6q23.2 (arginase) |
| Autosomal Dominant Ataxias | Neurologic Phenotype (Gait ataxia is a constant feature) |
Locus/Gene |
|---|---|---|
| SCA1 | Peripheral neuropathy Pyramidal signs Ophthalmoparesis |
6p22.3 ATXN1 |
| SCA2 | Slow saccades Facial fasciculation Hyporeflexia Dementia Peripheral neuropathy Extrapyramidal findings |
12q24.12 ATXN2 |
| SCA3 | Pyramidal and extrapyramidal signs Opthalmoplegia Eyelid retraction Amyotrophy Peripheral neuropathy |
14q32.12 ATXN3 |
| SCA4 | Sensory axonal neuropathy Pyramidal signs |
16q22.1 |
| SCA5 | Early onset, relatively pure cerebellar ataxia with dysarthria Slow progression |
11p13.2 SPTBN2 |
| SCA6 | Slow onset pure cerebellar ataxia with dysarthria, nystagmus | 19p13.13 CACNA1A |
| SCA7 | Vision loss - macular or retinal degeneration Dysarthria |
3p14.1 ATXN7 |
| SCA8 | Hyperreflexia, spasticity Impaired vibration sense |
13q21 ATXN8 |
| SCA9 | Ophthalmoplegia Dysarthria Pyramidal and extrapyramidal tract signs Weakness Posterior column signs |
Unknown |
| SCA10 | Pure cerebellar syndrome Seizures Dementia and dysphagia rarely |
22q13.31 ATXN10 |
| SCA11 | Slowly progressive mild ataxia |
15q15.2 TTBK2 |
| SCA12 | Tremor at onset Hyperreflexia Late dementia |
5q32 PPP2R2B |
| SCA13 | Childhood onset Associated cognitive and motor delays |
19q13.33 KCNC3 |
| SCA14 | Axial myoclonus Eye movement abnormalities |
19q13.42 PRKCG |
SCA15/SCA 16 |
Pure ataxia with slow progression Postural tremor Gaze palsy |
3p26.1 ITPR1 |
SCA17 |
Cognitive impairment Psychosis Seizures Huntington disease-like presentation Chorea |
6q27 TBP |
SCA18 |
Dysmetria Sensory axonal neuropathy Muscle weakness & atrophy Pes cavus |
7q22-q32 |
SCA19/22 |
Slowly progressive ataxia Hyporeflexia Cognitive decline Myoclonus |
1p13.2 KCND3 |
SCA20 |
Dysarthria and dysphonia Palatal tremor Spasmodic coughing |
11q12 |
| SCA21 | Cognitive disorders Extrapyramidal features Dysarthria Can have childhood onset |
1p36.33 TMEM240 |
SCA23 |
Sensory loss Vibration loss Tremor Memory deficits after 50 |
20p13 PDYN |
| SCA25 | Childhood onset most common Sensory neuropathy Gastrointestinal symptoms Nystagmus Facial tics |
2p21-p13 |
| SCA26 | Dysarthria Ocular pursuit abnormalities |
19p13.3 EEF2 |
SCA27 |
Tremor then gait and limb ataxia Behavioral outbursts Red-green colorblindness Strabismus |
13q33.1 FGF14 |
| SCA28 | Ophthalmoparesis Dysarthria Nystagmus Ptosis |
18p11.21 AFG3L2 |
| SCA29 | Congenital or childhood onset Nonprogressive ataxia Cognitive impairment Frequent falls Some motor symptoms can improve with age |
3p26.1 ITPR1 |
| SCA30 | Pure cerebellar syndrome Dysarthria Lower limb hyperreflexia |
4q34.3-q35.1 |
SCA31 |
Slowly progressive gait and limb ataxia Hearing loss Cerebellar dysarthria |
16q21 BEAN |
SCA32 |
Variable cognitive impairment, and azoospermia in males | 7q32-q33 |
| SCA34 | Papulosquamous erythematous plaques | 6q14.1 ELOVL4 |
| SCA35 | Upper limb involvement Torticollis |
20p13 TGM6 |
| SCA36 | Truncal instability Dysarthria Dysphagia Tongue atrophy or fasciculation Eye movement abnormalities Hearing loss |
20p13 NOP56 |
| SCA37 | Late onset falls and clumsiness Dysarthria Abnormal vertical eye movements |
1p32.2 DAB1 |
| SCA38 | Polyneuropathy Nystagmus Slow saccades |
6p12.1 ELOVL5 |
| SCA39 | Strabismus Saccadic pursuit dysfunction Horizontal gaze palsy Mild intellectual disability |
11q21-11q22.3 |
| SCA40 | Dysarthria Ocular dysmetria Tremor |
14q32.11-q32.12 CCDC88C |
| SCA41 | Late onset imbalance and gait ataxia | 4q27 TRPC3 |
| SCA42 | Dysarthria Saccadic eye movements Diplopia Nystagmus Spasticity |
17q21.33 CACNA1G |
| SCA43 | Slowly progressive gait and limb ataxia Peripheral neuropathy Mild atrophy of lower limbs |
3q25.2 MME |
| SCA44 | Frequent falls Dysarthria Dysphagia Dysmetria |
6q24.3 GRM1 |
| SCA45 | Relatively pure cerebellar syndrome Downbeat nystagmus Dysarthria |
5q33.1 FAT2 |
| SCA46 | Sensory neuropathy Cerebellar dysarthria |
19q13.2 PLD3 |
| SCA47 | Dysmetria Dysarthria Diplopia |
1p35.2 PUM1 |
| SCA48 | Cerebellar signs Cognitive impairment Anxiety |
16p13.3 STUB1 |
| CANPMR | Intellectual disability Hypotonia Dysarthria Dysmorphic facial features |
1p36.31-p36.23 CAMTA1 |
| ADCADN | Narcolepsy Sensorineural deafness Dementia |
19p13.2 DNMT1 |
| Dentatorubropallidoluysian atrophy (DRPLA) | Chorea Seizures Myoclonus Dementia |
12p13.31 ATN1 |
| *Gait ataxia is a constant feature |
| Disorder/Syndrome
|
Neurologic Phenotype | Inheritance | Locus/ Gene |
| Ataxia with selective vitamin E deficiency | Chronic ataxia | Autosomal recessive | 8q12.3 TTPA |
| Friedreich ataxia | Progressive ataxia plus | Autosomal recessive | 9q13-q21.11 FXN |
| Abetalipoproteinemia | Retinal degeneration Progressive ataxia Peripheral neuropathy |
Autosomal recessive | 4q24 MTTP |
| Hypobetalipoproteinemia | Retinal degeneration Progressive ataxia Peripheral neuropathy |
Autosomal codominant | 2q24 APOB |
| *Listed here due to overlap of clinical features with abetalipoproteinemia. | |||
Disorder/Syndrome |
Neurologic Phenotype |
Inheritance |
Gene Locus |
Gene Product/Biochemical Defect |
Cockayne syndrome type A |
Progressive ataxia plus Early onset severe syndrome |
Autosomal recessive |
5q11 |
ERCC8 |
Cockayne syndrome type B |
Progressive ataxia plus Classical type |
Autosomal dominant |
10q11-q21 |
ERCC6 |
Xeroderma pigmentosum |
Progressive ataxia plus |
Autosomal recessive |
Genetically heterogeneous with several complementation groups identified 9q34 locus (A) Other complementation groups involved are 2q21 (B & CS); 3p25.1 (C); 19q13.2(D); Unknown (E); 16p13 (F); 13q32-33 (G & CS) |
Mutations result in either defective damage-specific DNA-binding protein or defective excision repair (ERCC) Neurologic manifestations beginning in childhood relate to complementation group |
Ataxia Telangiectasia |
Progressive ataxia plus |
Autosomal recessive |
11q22-q23 |
ATM gene Product belongs to the P-13 kinase family of proteins involved in DNA damage recognition |
Ataxia with oculomotor apraxia type 1 (AOA1) |
FRDA-like hypoalbuminemia |
Autosomal recessive |
9p13.3 |
Aprataxin (APTX) Role in single-stranded DNA repair |
Ataxia with oculomotor apraxia type 2 (AOA2) Changed to autosomal recessive cerebellar ataxia (SCAR1) |
Ocular apraxia is an inconsistent feature. Ataxia Distal amyotrophy Peripheral neuropathy |
Autosomal recessive |
9q34 |
Senataxin (SETX) Protein involved in RNA maturation and termination |
Disorder/Syndrome |
Neurologic Phenotype |
Inheritance |
Gene Locus |
Gene Product/Biochemical Defect |
Autosomal recessive spastic ataxia of Charlevoix-Saguenay |
Chronic ataxia Spasticity Retinal abnormalities |
Autosomal recessive |
13q11 |
SACS gene codes for sacsin, which is involved in chaperone-mediated protein folding |
Leukoencephalopathy with VWM |
Progressive ataxia Spasticity Optic atrophy Seizures |
Autosomal recessive |
3q27 |
Mutations affect eIF2B |
4H syndrome |
Short stature Slowly progressive ataxia Hypogonadism Hypomyelination hypodontia |
Autosomal recessive |
Not known |
Not known |
| Disorder/Syndrome | Neurologic Phenotype | Inheritance | Locus/Gene |
|---|---|---|---|
| Refsum disease | Some have ataxia Retinitis pigmentosa Peripheral neuropathy Cardiac dysfunction Deafness |
Autosomal recessive | 10p13 PHYH |
| Cerebrotendinous xanthomatosis | Cerebellar ataxia Spinal cord involvement Pseudobulbar palsy Cataracts Palatal myoclonus Seizures Mental decline Tendon xanthomas |
Autosomal recessive | 2q35 CYP27A1 |
| Biotinidase deficiency | Intermittent ataxia Sensorineural hearing loss Seizures Developmental delay Skin rashes, alopecia Hearing loss Optic atrophy |
Autosomal recessive | 3q25.1 BTD |
| L-2 hydroxyglutaric acidemia | Intellectual disability Seizures Progressive ataxia and other cerebellar signs Macrocephaly |
Autosomal recessive | 14q21.3 L2HGDH |
| Succinic-semialdehyde dehydrogenase deficiency | Ataxia Seizures Hypotonia Developmental delay of motor, mental and language skills |
Autosomal recessive | 6p22.3 ALDH5A1 |
Late infantile and juvenile sphingolipidoses 1. Metachromatic leukodystrophy 2. Krabbe disease 3. Gaucher type III 4. Niemann-Pick C disease 5. GM2 gangliosidosis |
Progressive ataxia plus Seizures Psychomotor regression Spasticity Extrapyramidal features Supranuclear gaze palsies |
Autosomal recessive | 1. 22q13.3 -ARSA 2. 14q31.3- GALC 3. 1q22- GBA 4. 18q11.2– NPC1 5. 5q33.1- GM2A |
| Congenital disorders of glycosylation type Ia | Psychomotor retardation Axial hypotonia Abnormal eye movement Peripheral neuropathy Cerebellar ataxia |
Autosomal recessive | 16p13.2 PMM2 |
| Marinesco-Sjögren syndrome | Ataxia Congenital cataract Hypotonia Myopathy Delay of psychomotor development |
Autosomal recessive | 5q31.2 SIL1 |
Disorder/Syndrome |
Neurologic Phenotype |
Inheritance |
Gene Product/Biochemical Defect |
NARP syndrome |
Progressive ataxia plus |
Maternal inheritance |
Mitochondrial ATP 6 NARP 8993 mutation causing base substitution T-G or T-C at nucleotide position 8993 |
Leigh disease |
Progressive ataxia plus Lactic acidosis |
Autosomal recessive/maternal inheritance |
Multiple biochemical and molecular defects underlie the condition, eg, PDHC deficiency, cytochrome oxidase C deficiency, mitochondrial ATPase 6 |
CoQ-10 responsive ataxia |
Progressive ataxia in childhood Developmental delay Seizures Cerebellar atrophy on MRI |
Autosomal recessive |
Mutations in the gene CABC1 or ADCK3 are described. The gene codes for a putative protein kinase associated with ubiquinone biosynthesis. |
Type |
Unverricht-Lundborg syndrome |
Neurologic Phenotype |
Inheritance |
Locus |
Dodecamer repeat expansion |
Unverricht-Lundborg syndrome |
Myoclonus Ataxia Seizures |
Autosomal recessive 21q22.2 |
21q22.2 CSTB |
Inherited enzyme defect |
Lafora body disease |
Myoclonus Ataxia Seizures Mental decline |
Autosomal recessive 2A- 6q24.3 EPM2A |
2B – 6p22.3 NHLRC1 |
Inherited enzyme defect |
Neuronal ceroid lipofuscinosis |
Myoclonus Ataxia Seizures Visual impairment |
Autosomal recessive |
11p15.4 TPP1 |
Mitochondrial cytopathy |
MERRF |
Myoclonic epilepsy Ataxia Hearing disturbance Mental deterioration Muscle atrophy |
Maternal inheritance |
Multiple mutations, including: MTTK, MTTL1, MTTH, MTTS1, MTTS2, MTTF, MTND5 |

What would you like to print?
- Background
- Molecular Genetics and Putative Mechanisms of Cerebellar Disease
- Nonprogressive Cerebellar Ataxias
- Intermittent or Episodic Ataxias
- Chronic or Progressive Ataxias
- Progressive Ataxias With Polymyoclonus and Epileptic Seizures
- Other Disorders
- Approach to Patients With a Suspected Inherited Ataxia
- Show All
- Media Gallery
- Tables
- References




