Background
The term epileptic encephalopathy describes a heterogeneous group of epilepsy syndromes associated with severe cognitive and behavioral disturbances. These disorders vary in their age of onset, developmental outcome, etiologies, neuropsychological deficits, electroencephalographic (EEG) patterns, seizure types, and prognosis, but all may have a significant impact on neurological development. [1, 2]
In 2001, the International League Against Epilepsy (ILAE) Task Force on Classification and Terminology proposed a modified diagnostic scheme for epileptic seizures and epilepsy that, for the first time, recognized epileptic encephalopathies as a distinct category. [3, 4]
The ILAE defined an epileptic encephalopathy as a condition in which "the epileptiform EEG abnormalities themselves are believed to contribute to a progressive disturbance in cerebral function."
Later in 2010, researchers defined epileptic encephalopathy as a condition where the epileptic activity itself may contribute to severe cognitive and behavioral impairments above and beyond what might be expected from the underlying pathology alone (e.g., cortical malformation), and that these can worsen over time. [5]
This category includes the following epilepsy syndromes:
-
Early myoclonic encephalopathy (EME)
-
Early infantile epileptic encephalopathy (EIEE/Ohtahara syndrome)
-
Dravet syndrome (severe myoclonic epilepsy in infancy; SMEI)
-
Malignant epilepsy with migrating partial seizures in infancy
-
Doose syndrome (myoclonic astatic epilepsy)
-
Myoclonic status in nonprogressive encephalopathies
-
Epilepsy with continuous spike-waves during slow sleep (CSWS)
-
Rasmussen's encephalitis
Because this concept is evolving, this listing is not definitive; other epilepsy syndromes, such as some cases of benign focal childhood epilepsy with centro-temporal spikes (benign rolandic epilepsy) and autism with epileptiform EEG abnormalities, may also fit under this rubric. [6] These 2 conditions are not currently considered epileptic encephalopathies, but there is increasing evidence that epilepsy or epileptiform activity may contribute to encephalopathy in a subset of cases.
"Epileptic encephalopathy" is the most commonly used phrase in the literature. Note that the term epileptic encephalopathy may refer to conditions with severe and frequent ictal EEG activity (actual seizures) as a more prominent component. In contrast, the term epileptiform encephalopathies describes those conditions in which the interictal epileptiform EEG abnormalities may be more prominent than the clinical seizures.
Chatrian et al in their 1974 glossary of EEG terms defined the term epileptiform to describe distinct waves or complexes, distinguishable from the background activity, which resemble the waveforms recorded in a proportion of human subjects suffering from an epileptic disorder. [7] Epileptiform patterns include spike and sharp wave discharges, either alone or accompanied by slow waves, occurring singly or in bursts lasting at most a few seconds (see image below).
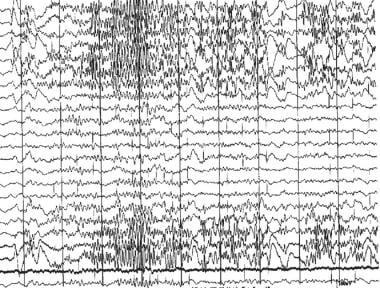 Epileptic and epileptiform encephalopathies. EEG showing an epileptiform beta frequency burst.
Epileptic and epileptiform encephalopathies. EEG showing an epileptiform beta frequency burst.
In clinical practice, the ability to distinguish between epileptiform activity and an epileptic disorder may be challenging, as variability may be seen within each epilepsy syndrome and within a given child over time. However, in some cases, severe developmental regression may be seen in the context of few seizures but severe interictal epileptiform abnormalities, as in some cases of LKS).
The quantity of epileptiform activity does not correlate well with seizure severity. However, observational and anecdotal evidence correlate the quantity of epileptiform activity with the degree of cognitive impairment. Additional well-designed studies are needed to sufficiently quantify and correlate interictal epileptiform activity with neuropsychological and developmental measures.
Inherent in the notion of an epileptic encephalopathy is that limiting or suppressing EEG ictal and/or interictal activity may improve cognitive and behavioral outcome. Anecdotal and small series data support this concept; however, this remains controversial due to the lack of data from larger well-designed studies.
A common feature is that these disorders are usually refractory to standard antiepileptic drugs (AEDs). As a result, more aggressive use of AEDs considered effective in suppressing interictal epileptiform discharges (eg, benzodiazepines, valproic acid, lamotrigine), immunomodulatory therapies (eg, corticosteroids, intravenous immunoglobulin [IVIG], plasmapheresis), ketogenic diet, and surgical options are often considered. (See Treatment and Management, as well as Medications.)
At this time, it remains unclear how much of the dysfunction seen in these disorders is due to epileptiform EEG activity and how much is due to the underlying cause of the epilepsy syndrome. Consequently, a useful guideline is to "treat the patient, not the EEG." Data are insufficient to recommend treatment for the sole purpose of minimizing interictal epileptiform activity at this time, especially when the clinical symptoms of the epileptiform activity on the patient are unclear.
For more information, see Epilepsy and Seizures and First Pediatric Seizure.
Terminology and definitions
Epilepsy is a chronic condition with spontaneous, recurrent seizures; a seizure is defined as a clinical event associated with a transient, hypersynchronous neuronal discharge.
Epileptic denotes the presence of epilepsy.
Epileptic seizure is a clinical event associated with a transient, hypersynchronous neuronal discharge and represents only the symptom of a potential underlying brain pathology, not the actual disease.
Encephalopathy refers to a disturbance in brain functioning, particularly in intellectual activity or higher cortical functioning as used in this review.
Epileptiform refers to spike waves, sharp waves, spike and wave activity, or other rhythmic waveforms that imply epilepsy or may be associated with epilepsy. However, epileptiform activity alone does not confirm a diagnosis of epilepsy.
Epileptic or epileptiform encephalopathy is a category of severe epilepsy syndromes of infancy or early childhood, in which the epileptiform EEG abnormalities themselves are believed to contribute to a progressive disturbance in cerebral function.
More precisely, the term epileptic encephalopathies may be used to refer to those syndromes characterized by very frequent seizures, where as epileptiform encephalopathies may be used to refer to those syndromes that generally occur later and where the EEG epileptiform activity is more prominent than clinical seizures.
Although the term epileptic aphasia has been used for LKS, epileptic aphasia by its strict definition refers to an aphasia caused by an actual seizure or, in other words, an ictal aphasia.
Epileptiform aphasia refers to a language disorder—expressive, receptive, or mixed— associated with epileptiform features on EEG. The terms congenital aphasia, developmental aphasia, or acquired aphasia are used with this to describe whether the condition is developmental or acquired. Acquired aphasia implies previously normal language development with subsequent regression. Note that regression might occur even in developmental language disorders or the congenital aphasias.
Pathophysiology
The epileptic encephalopathies are a group of age-specific epilepsy syndromes of diverse etiologies that share the potential for causing significant cognitive impairment. The underlying mechanisms of these disorders are still poorly understood.
Identifiable factors that may influence the course and degree of cognitive and behavioral impairment in these disorders include the following:
-
Underlying etiology
-
Age of onset of epilepsy
-
Seizure frequency and severity
-
Interictal epileptiform activity severity
-
Treatment-related adverse effects
-
Cumulative detrimental effects of severe chronic epilepsy
-
Genetic factors
Electrical dysfunction
It remains unclear how much electrical dysfunction contributes to the neuropsychological impairments seen in these disorders. Frequent seizures and/or interictal discharges may significantly disrupt the function of neuronal networks involved in language, learning, memory, behavioral regulation, and other higher cortical functions, resulting in either transient or permanent deficits. For example, continuous abnormal discharges during sleep may cause disruption of hippocampal function and interfere with learning and memory while awake and memory consolidation in sleep. [8, 9]
The deficits seen in some epileptic encephalopathies appear to generally correlate with the location, frequency, and degree of spread of abnormal electrical activity, as in LKS and CSWS; however, further studies are required to better quantify and characterize the evolution of these deficits with the various potential contributing factors. This characterization is complicated by the fact that evaluating subtle cognitive impairments from the involvement of noneloquent cortex requires testing of performance.
The duration of electrical dysfunction may in part determine the severity of the disorder.
Impairment at the exact moment of an interictal discharge has been described and is termed transient cognitive impairment. [10, 11, 12, 13, 14, 15, 16] Although challenging to demonstrate, this phenomenon appears to be due to a temporary disruption of a cortical network involved in a particular function at the time of an interictal epileptiform discharge.
Longer-duration dysfunction may be seen during ictal and postictal states, which may last from minutes to days, depending in part on the severity of the seizure and the patient’s cognitive reserve. More chronic, potentially reversible dysfunction may also be seen, such as in the subset of children with benign focal epilepsy of childhood with centro-temporal spike discharges (BECTS) who demonstrate a variety of neuropsychological deficits that may be reversible. [17, 18, 19]
More chronic and permanent impairment may be seen in more severe disorders, such as LGS. The more severe epileptic encephalopathies fall into this category.
Epileptiform activity during sleep
In epileptic or epileptiform encephalopathies, ictal and/or interictal epileptiform activity often becomes more frequent during sleep. [20] When discharges are present in the awake state, this is termed sleep potentiation. If discharges are present only during sleep, this is termed sleep activation.
The role of sleep activation, particularly in electrical status epilepticus of sleep (ESES), offers an appealing and challenging paradigm that could lead to better understanding of the pathophysiologic basis of these conditions. Two crucial questions, as follows, still await an answer:
-
What are the mechanisms involved in the generation of such significant, interictal, sleep activation?
-
What are the mechanisms involved in the cognitive/developmental regression that accompanies these conditions?
Though still unclear, evidence suggests that defective mechanisms of synaptogenesis and thalamocortical circuit formation during a critical period may be involved in the generation of CSWS. Secondary bilateral synchrony, which is facilitated by the corpus callosum and that may involve the thalamocortical connections, was hypothesized as the possible mechanism for the generation of ESES discharges. [21, 22, 23]
Thalamic injury
An association between CSWS and early thalamic injury has been reported. A review of EEG abnormalities in 32 children with early thalamic injury, primarily due to vascular mechanisms, revealed that 29 out of the 32 patients showed significant sleep activation. [24]
Among these 29 patients, 2 different groups were distinguished: the first included typical CSWS (12 cases), generally with symmetry of spike and waves and often with no spindle at all. Patients in the second group had a typical asymmetry of spike and waves and the presence or reduction of spindles, plus other atypical features concerning synchronism and morphology of spike and waves. [24]
Behavioral disorders were significantly more present in patients with a true CSWS; their improvement paralleled the disappearance of CSWS. Generally, the predominant injury was in the lateral aspect of the thalamus including reticular nucleus and ventral nuclei. [24]
A case-control study was performed on early developmental lesions in children with clinical presentation consistent with CSWS, and prominent sleep-potentiated epileptiform activity. The study compared 100 children with such prominent sleep potentiated epileptiform activity (>50% spike percentage) to 47 children without such EEG findings. The children who had the prominent sleep potentiated spikes had a higher rate of having early brain injury, in particular early thalamic injury. [25]
Etiology
The causes of epileptic encephalopathy vary among the different syndromes.
Early myoclonic encephalopathy
The etiology is often unknown. Metabolic disorders, including nonketotic hyperglycinemia, have been described in early myoclonic encephalopathy and should be pursued. Structural lesions are rare.
Infantile spasms (West syndrome)
No clear etiology is found in approximately 40% of cases. [26] There is a broad range of potential causes, including cerebral malformations, infection, hemorrhage, hypoxic-ischemic injury, metabolic disorders, and genetic conditions (eg, Down syndrome).
Malignant epilepsy with migrating partial seizures in infancy
In most cases, there is no clear etiology or structural problems, suggesting genetic factors may be causative or contributory.
Severe myoclonic epilepsy of infancy (Dravet syndrome)
Most cases are associated with various mutations in the sodium channel gene SCN1A. Mutations in SCN1A may also be seen in other conditions; thus, it is not a specific finding. Neuroimaging is either normal or reveals nonspecific abnormalities. [27]
Myoclonic status in nonprogressive encephalopathies
A genetic cause is identifiable in approximately half of children, including Angelman syndrome and 4p- syndrome. [28] Other reported causes include hypoxic-ischemic injury and cortical dysplasia.
Myoclonic-astatic epilepsy (Doose syndrome)
Most cases are idiopathic with normal neuroimaging. A genetic etiology has been hypothesized given it sometimes has an association with febrile seizures and GEFS+; however, no specific gene has been implicated.
Lennox-Gastaut syndrome (LGS)
A broad range of acquired and developmental etiologies have been described, including cerebral malformations, encephalitis, and hypoxic-ischemic injury. [29]
Landau-Kleffner syndrome and epilepsy with continuous spikes-waves during slow sleep
Most cases of Landau-Kleffner syndrome are idiopathic, with normal results on neuroimaging; however, volumetric MRI analysis has revealed decreased volume in bilateral superior temporal gyrus and planum temporale in studied cases. [30] Symptomatic cases of LKS and CSWS are described and are likely more common in CSWS.
Benign childhood epilepsy with centro-temporal spike discharges (benign rolandic epilepsy)
A genetic etiology is suspected, and recent work has implicated that mutation of the Elongator Protein Complex 4 may confer genetic susceptibility. [31]
Autistic regression with epileptiform EEG findings
Epilepsy may aggravate autistic symptoms and interfere with developmental progress, independent of autism in some children; however, it is unclear if it is causative. In some conditions (eg, tuberous sclerosis, LKS), children may have some autistic features, though they usually do not meet full criteria for autism over time.
Proposed explanations for the coexistence of autism and epilepsy include the following:
-
They are independent conditions
-
The same underlying pathology results in an autistic phenotype and epilepsy
-
An epileptic process in early development impair the normal formation of networks involved in social skills and communication
-
A focal brain lesion affecting the frontal or limbic system may result in an autistic phenotype and the potential for epilepsy
-
Epilepsy may result in cognitive dysfunction and an "autistic withdrawal" in "vulnerable" children [32]
Epidemiology
In a 20-year epidemiological study of childhood epilepsy syndromes from Tel Aviv, Kramer et al reported the following distribution of epileptic encephalopathy cases [33] :
-
West syndrome - 9% of childhood epilepsy cases
-
Myoclonic seizures - 2.2%
-
Lennox-Gastaut syndrome - 1.5%
-
LKS, Ohtahara syndrome, myoclonic astatic epilepsy, and ESES - 0.2% each
Each of these childhood epilepsy syndromes has its own characteristic age of onset.
Prognosis
The prognosis is related to the underlying disorder. The severity of developmental impairment varies with the type of epilepsy.
Early infantile epileptic encephalopathy (Ohtahara syndrome)
The prognosis is very poor. Most children either die or are severely neurologically impaired. All surviving children have global developmental delays. Some children may progress to West syndrome and Lennox-Gastaut syndrome. These 3 disorders are considered to be on a spectrum by some authors. Progression to hypsarrhythmia portends a poorer prognosis.
Early myoclonic encephalopathy
The prognosis is poor. Neurological abnormalities are common, and most children have minimal developmental progress. Reported mortality is a high as 50% during the first year of life. [34]
Infantile spasms (West syndrome)
Development remains unaffected only in a minority. Most children experience slowing, plateauing, or regression of their developmental trajectory. An extensive literature review revealed that 16% of patients had normal development and 47% had continued seizures at an average follow-up of 31 months. [26] No specific AED has been shown to affect long-term developmental outcome.
The developmental prognosis partially depends on the etiology. When classified by etiology, normal development was described in 51% of cryptogenic cases versus only 6% of symptomatic cases. Approximately 17% of cases evolved into Lennox-Gastaut syndrome.
Malignant epilepsy with migrating partial seizures in infancy
Developmental regression is common. Death has been reported in infancy and childhood in severe cases.
Severe myoclonic epilepsy of infancy (Dravet syndrome)
Development is normal initially, followed by regression occurring by the second or third year of life and progressing to a significant intellectual disability. Cognition is severely affected, and most patients have motor and coordination dysfunction. In a series of patients followed into adulthood, approximately half had an IQ below 50. [35] Seizures continue into adulthood, and mortality increases from epilepsy-related causes.
Myoclonic status in nonprogressive encephalopathies
Affected children have a poor prognosis, experiencing developmental regression, and eventual severe intellectual disability. The repeated episodes of myoclonic status may contribute to cognitive deterioration.
Myoclonic-astatic epilepsy (Doose syndrome)
The prognosis is variable and difficult to predict. After several years, the seizures may remit in 54-89% of patients. [36] The cognitive outcome ranges from no sequelae in most cases to progressive cognitive impairment in a minority. Approximately 18% may have a poor cognitive outcome. [37]
A family history of epilepsy and recurrent episodes of status epilepticus may portend a worse prognosis. Epilepsy longer than 3 years’ duration and nocturnal tonic seizures, characteristic of Lennox-Gastaut syndrome, may also suggest a worse prognosis in some patients.
Lennox-Gastaut syndrome (LGS)
The developmental outcome is poor. Symptomatic Lennox-Gastaut syndrome increases the risk of intellectual disability, which is reported in up to 100% of symptomatic cases in long-term follow-up. [38] Other factors increasing the risk of intellectual disability include earlier age of onset and history of infantile spasms. Most patients continue having seizures. Early remission of epilepsy does not necessarily improve cognitive outcome.
Landau-Kleffner syndrome and epilepsy with continuous spikes-waves during slow sleep
The prognosis is variable. The epilepsy and ESES pattern improve and may remit after several years, whereas most children are left with varying degrees of language and cognitive dysfunction. Neuropsychological assessments should be performed in order to gauge developmental progress and the effect of treatments over time.
Benign childhood epilepsy with centro-temporal spike discharges (benign rolandic epilepsy)
Most children are developmentally normal and do not exhibit any obvious problems. However, a subset of children does experience cognitive impairment.
An abundance of literature on benign rolandic epilepsy (BRE) has described a variety of neuropsychological deficits, but with no uniform profile of impairment identifiable and variable study methodologies. Bilateral rolandic EEG discharges are associated with poorer cognitive function than unilateral discharges. Left hemisphere discharges have also been associated with verbal problems, while right hemisphere discharges have been associated with nonverbal difficulties.
Unfortunately, few studies have attempted to correlate EEG abnormalities, including spike discharge frequency, with neuropsychological deficits. EEG findings that have been correlated with cognitive problems include a high awake or sleep spike index and intermittent EEG slowing. [19, 39, 40, 41, 42, 43] However, further investigations are needed to clarify these relationships and better define this aspect of this syndrome.
Patient Education
Input from a neurologist, developmental pediatrician, psychologist, neuropsychologist, audiologist, or speech pathologist is needed to determine the proper educational program.
For patient education information, see the Brain and Nervous System Center, as well as Epilepsy.
-
Epileptic and epileptiform encephalopathies. EEG showing an epileptiform beta frequency burst.
-
EEG of a patient with Landau-Kleffner syndrome showing electrical status epilepticus of sleep (ESES).
-
Epileptic and epileptiform encephalopathies. Waking EEG in Landau-Kleffner syndrome, showing left posterior spikes.
-
Epileptic and epileptiform encephalopathies. EEG in Landau-Kleffner syndrome (LKS), before and after treatment with prednisone. The left EEG tracing shows electrical status epilepticus of sleep. The right tracing, obtained after 6 months of prednisone treatment, is normal.
-
Epileptic and epileptiform encephalopathies. Frequency-modulated auditory evoked response (FMAER), before and after treatment with prednisone. The left FMAER is absent. The right FMAER is normal following treatment.
Tables

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Approach Considerations
- Early Infantile Epileptic Encephalopathy (Ohtahara Syndrome)
- Early Myoclonic Encephalopathy
- Infantile Spasms (West Syndrome)
- Malignant Epilepsy with Migrating Partial Seizures in Infancy
- Severe Myoclonic Epilepsy of Infancy (Dravet Syndrome)
- Myoclonic Status in Nonprogressive Encephalopathies
- CDKL5 Deficiency Disorder
- Surgical Care
- Consultations
- Show All
- Medication
- Media Gallery
- References





