Overview of Neuroprotective Agents
An ischemic stroke occurs when a cerebral vessel occludes, obstructing blood flow to a portion of the brain (see the image below). Currently, the only approved medical therapy for acute ischemic stroke is tissue plasminogen activator (tPA), a thrombolytic agent that targets the thrombus within the blood vessel. [1, 2] For more information, see Ischemic Stroke.
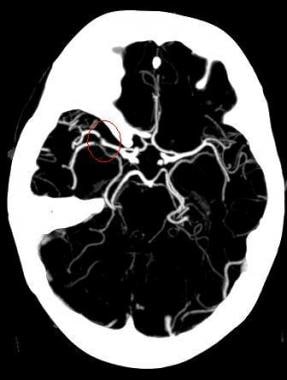 MIP image from a CTA demonstrates a filling defect or high-grade stenosis at the branching point of the right MCA trunk (red circle), suspicious for thrombus or embolus. CTA is highly accurate in detecting large vessel stenosis and occlusions, which account for approximately one-third of ischemic strokes.
MIP image from a CTA demonstrates a filling defect or high-grade stenosis at the branching point of the right MCA trunk (red circle), suspicious for thrombus or embolus. CTA is highly accurate in detecting large vessel stenosis and occlusions, which account for approximately one-third of ischemic strokes.
Despite the availability of thrombolytic therapy, stroke is the fifth leading cause of death in the United States and a leading cause of adult disability. To reduce the devastating impact of stroke on society, physicians continue to seek ways to achieve better functional recovery in stroke patients. Neuroprotective agents, another approach to stroke treatment, have generated long-term interest.
Neuroprotective agents are used in an attempt to save ischemic neurons in the brain from irreversible injury. [3] Studies in animals indicate a period of at least 4 hours after onset of complete ischemia in which many potentially viable neurons exist in the ischemic penumbra (ie, the rim of the infarct). In humans, the ischemia may be less complete, and the time window may be longer, but human patients also tend to be older, with comorbidities that may limit benefit.
Because many neuroprotective drugs reduce ischemic damage in animal models of stroke, this line of pharmaceutical research hold promise despite the lack of efficacy in human models to date. Many investigators are searching for a safe agent that can limit ischemic damage in human stroke.
One action of neuroprotective agents limits acute injury to neurons in the ischemic penumbra. Neurons in the penumbra are less likely to suffer irreversible injury at early time points than are neurons in the infarct core. Many of these agents modulate neuronal receptors to reduce release of excitatory neurotransmitters, which contribute to early neuronal injury.
Other neuroprotective agents prevent potentially detrimental events associated with return of blood flow. Although return of blood flow to the brain is generally associated with improved outcome, reperfusion may contribute to additional brain injury. [4] Returning blood contains leukocytes that may occlude small vessels and release toxic products.
This article discusses 2 types of neuroprotective agents, one that prevents early ischemic injury and another that prevents reperfusion injury. Clinical trial results of these agents are summarized.
Prevention of Early Ischemic Injury
N-methyl-D-aspartate receptor antagonists
Ischemia leads to excessive activation of excitatory amino acid receptors, accumulation of intracellular calcium, and release of other toxic products that cause cellular injury. By preventing excitatory neurotransmitter release, neuroprotective agents may reduce deleterious effects of ischemia on cells.
The most commonly studied neuroprotective agents for acute stroke block the N-methyl-D-aspartate (NMDA) receptor. Dextrorphan, a noncompetitive NMDA antagonist and a metabolite of the cough suppressant dextromethorphan, was the first NMDA antagonist studied in human stroke patients. [5] Unfortunately, dextrorphan caused hallucinations and agitation; it also produced hypotension, which limited its use. [5]
Selfotel, a competitive NMDA antagonist, showed trends toward higher mortality in treated patients than in placebo-treated cohorts; thus, trials were stopped prematurely. [6] A trial of another NMDA receptor antagonist, aptiganel HCl (Cerestat), was terminated because of concerns regarding benefit-to-risk ratios. [7]
In patients treated with NMDA antagonists, the adverse effects of hallucinations and agitation mimic those seen with phencyclidine, which binds at a similar site. To avoid these phencyclidine-like effects, indirect NMDA receptor antagonists that work at the glycine site of the receptor were developed. These agents prevent glycine from binding, which in turn prevents glutamate from activating the receptor. Early clinical studies suggested that psychomimetic side effects occur less frequently with these glycine-site NMDA antagonists. [8]
A large efficacy trial using the agent GV150526 on 1367 patients was completed in 2000. Although the drug was reported to be safe and well tolerated, no improvement was observed in any of the 3-month outcome measures. [9] No further trials are planned.
Research in NMDA modulation in stroke continues, however. Using a pretreatment strategy in a novel "human embolic stroke model" NA-1, a neuroprotectant that may reduce NMDA mediated injury by disrupting down-stream pathways was associated with a reduction in MRI-based infarcts in a recent clinical trial. [10]
Magnesium
Magnesium is another agent with actions on the NMDA receptor and a low incidence of adverse effects. It may reduce ischemic injury by increasing regional blood flow, antagonizing voltage-sensitive calcium channels, and blocking the NMDA receptor. [11] In myocardial infarction and small stroke studies, patients tolerated the drug. [12]
While a phase III trial investigating the efficacy of magnesium in stroke, the Intravenous Magnesium Efficacy Study, that treated patients within 12 hours of symptom onset did not show efficacy, the long time window may have been to blame and magnesium remained of great interest. [13]
The Field Administration of Stroke Therapy - Magnesium Phase III (FAST-MAG) trial studied the delivery of magnesium in the field by paramedics to greatly reduce the time to treatment. The majority of patients in this 1700-patient trial were enrolled within 1 hour of symptom onset. The trial was the first to assess treatment with a neuroprotectant in the very early period after clinical ischemic stroke.
This study by Saver et al concluded that prehospital initiation of magnesium sulfate therapy as neuroprotection in acute stroke was safe and it was feasible to start therapy within 2 hours after the onset of stroke symptoms. However, the study found that the therapy did not improve disability outcomes at 90 days. [14]
Nalmefene
Modulating other non-NMDA receptors and channels also can reduce excitatory neurotransmitter release. Nalmefene (Cervene) is a narcotic receptor antagonist that reduces levels of excitatory neurotransmitters contributing to cellular injury in early ischemia. [15] Unlike NMDA receptor antagonists, this drug causes minimal side effects.
Post hoc analyses of early studies suggest that the drug may have more benefit in patients younger than 70 years. [16] However, no clinical benefit was found in a phase III clinical trial in which the drug was administered intravenously within 6 hours of symptom onset. [15] No further trials are planned.
Lubeluzole
The exact mechanism of action of lubeluzole, a drug effective in animal models, is unclear. The drug may block sodium channels in cells. In addition, it may reduce the release of nitric oxide, a neurotransmitter generated by activation of the NMDA receptor. Although stroke severity and patient age appeared to influence outcome in early studies, [17] a later trial was unable to confirm that lubeluzole was effective in these subsets of acute stroke patients [18] . Clinical stroke research with this drug has been abandoned.
Clomethiazole
Clomethiazole, a gamma-aminobutyric acid agonist, decreases excitatory neurotransmission by increasing activity of inhibitory pathways. [19] In Europe, clomethiazole's central nervous system inhibitory properties led to its use as an anticonvulsant and sedative.
The potential efficacy of clomethiazole as a neuroprotective agent in ischemia was first investigated in Europe as part of the Clomethiazole Acute Stroke Study. Patients received a 24-hour intravenous infusion of clomethiazole or placebo within 12 hours of symptom onset. [20] As predicted by the drug's inhibitory effects, its primary side effect was sedation.
Overall, the study result was negative. Functional outcome at 3 months, the primary endpoint, was not significantly better with clomethiazole than with placebo. In a subgroup of patients with large strokes, however, there was a 37% relative improvement.
A large phase III trial was then performed in 1198 patients, focusing on patients with large strokes, which were defined by the presence of higher cortical dysfunction, limb weakness, and visual field disturbances. [19] The results were negative, with 42% of the clomethiazole group and 46% of the placebo group showing a good outcome.
Other drugs
Calcium channel blockers were extensively evaluated in acute stroke with the hope that stemming excessive cellular calcium influx caused by ischemia might prevent neuronal injury. Studies of calcium channel blockers did not show efficacy in stroke treatment. A clinical trial that assessed the usefulness of oral nimodipine given within 6 hours of symptom onset was terminated early after analysis of the first 439 patients predicted no beneficial effect of the drug. [21]
Several drugs that modulate neurotransmission or ion channels by novel mechanisms have completed clinical trials. These drugs included an α-amino-3-hydroxy-5-methylisoxazole-4-proprionic acid antagonist developed by Yamanouchi, USA, Inc; a serotonin agonist, repinotan, developed by Bayer Corporation; and a transmembrane potassium channel modulator developed by Bristol-Myers Squibb.
Unfortunately, none of the clinical trials showed efficacy for the investigational treatment. The serotonin agonist, repinotan, was given early, after only 4.5 hours of symptom onset. ONO 2506, a novel neuroprotectant developed by Ono Pharmaceutical Co, Ltd, that inhibits astrocyte activation, was administered within 6 hours of stroke onset. A futility analysis performed in May 2005 led to discontinuation of the trial in the United States.
Free-radical scavenger tirilazad
Another target of neuroprotective therapy in acute ischemic stroke is free-radical generation, which leads to further release of calcium and excitatory neurotransmitters. The free-radical scavenger tirilazad did not show benefit in an acute stroke trial. [22] The drug also was investigated in subarachnoid hemorrhage and in traumatic brain injury, without convincing evidence of benefit.
Free-radical trapping NXY-059
The free-radical trapping agent NXY-059 showed efficacy in a single acute stroke treatment trial, but the positive results were not duplicated in a second trial. Stroke-Acute Ischemic NXY Treatment (SAINT I) was the first of 2 efficacy trials. [23] The drug was delivered intravenously over 72 hours in patients within 6 hours of stroke symptom onset, although a forced allocation scheme maintained an average treatment time of less than 4 hours. Patients were required to have limb weakness and a score of at least 6 on the National Institutes of Health Stroke Scale (NIHSS).
Patients who received treatment had significantly better outcomes on the primary endpoint, the distribution of scores on the modified Rankin scale that assessed disability at 90 days. The co-primary endpoint, change from baseline on the NIHSS, did not show a difference between treatment groups. Of interest, post hoc analyses in patients treated with tPA showed that patients who received NXY-059 and tPA had significantly fewer hemorrhagic transformations than those who received placebo and tPA, and symptomatic hemorrhagic transformations also occurred less frequently in this group.
The second efficacy trial, SAINT II, completed enrollment in the summer of 2006 and was reported in 2007. [24] Although the design mimicked that of SAINT I, again initiating treatment within 6 hours of symptom onset, the results showed no efficacy for the treatment arm compared with placebo and no difference in the rate of symptomatic or asymptomatic hemorrhages in the patients treated with NXY-059 and tPA.
A pooled analysis of SAINT I and SAINT II also did not show any difference between the treatment and control arms. [25] There was no decrease in the rate of symptomatic or asymptomatic hemorrhage in the group treated with NXY-059 compared with placebo for those who received tPA.
Other
A phase III trial investigated albumin versus placebo given within 5 hours of symptom onset [26] . In preclinical studies, albumin appears to have both antioxidant properties and the ability to increase blood flow to the penumbra. The trial, however, was stopped early for futility.
Hypothermia is being evaluated for its neuroprotective capabilities. [27] One study is evaluating hypothermia (treatment within 300 min) in conjunction with a combination of caffeine and ethanol (caffeinol; treatment within 240 min).
Prevention of Reperfusion Injury
Mechanism of reperfusion
Despite the good outcome generally associated with reopening a blood vessel, additional brain injury may result when reperfusion occurs. [4] When white blood cells reenter a previously hypoperfused region via returning blood, they can occlude small vessels, producing additional ischemia. In addition, leukocytes release toxic products that can lead to free radical and cytokine formation.
Agents that prevent white blood cells from adhering to vessel walls, limit formation of free radicals, or promote neuronal repair may protect the brain from additional injury during reperfusion. Neuroprotective agents that work primarily during reperfusion may have a longer window of therapeutic effect than drugs that work earlier in the ischemic cascade.
For related information, see Reperfusion Injury in Stroke.
Enlimomab
Monoclonal antibodies can block an intercellular adhesion molecule (ICAM) on the endothelium to prevent adhesion of white blood cells to the vessel wall. [28] Because anti-ICAM antibodies appear to block an early step in reperfusion-related injury, they present a hopeful mechanism for preserving neuronal function.
A large multicenter trial assessed clinical efficacy of anti-ICAM-1. [29] More than 600 patients received either IV boluses of murine monoclonal antibody to ICAM-1 (enlimomab) or placebo for 5 days, beginning within 6 hours after symptom onset.
Treated subjects were found to have higher mortality rates and worse outcomes than subjects in the placebo group. A marked increase in fevers occurred in patients who received enlimomab. Elevated temperatures had previously been found to worsen stroke outcome.
A viable explanation for the adverse effects seen in the anti-ICAM-1 trial is that the patients who were treated developed an immune response to the murine antibody. Although enlimomab was not clinically useful in the treatment of stroke, researchers hoped that its mechanism of action still could have a role in limiting neuronal injury.
Hu23F2G
A phase III trial was then done by using a human antileukocytic antibody, Hu23F2G, developed by ICOS Corporation. Because the antibody is humanized and not murine, the developers hoped that this agent would avoid the unwanted effects of enlimomab. This agent did not appear to produce the immune response seen with enlimomab. However, no clinical benefit was seen with Hu23F2G on any of the planned measures. No further studies with this agent are planned, though smaller antibodies that target specific leukocyte receptors may still have potential.
Tetracycline antibiotics
The tetracycline family of antibiotics has also been shown to reduce leukocyte infiltration and improve experimental stroke outcome. [30] A phase I dose-finding trial using minocycline to treat ischemic stroke (6-h window) was completed. [31] In addition to providing potential neuroprotection, minocycline also appears to decrease levels of matrix metalloproteinase-9, which has been associated with recombinant tissue plasminogen activator (rtPA)–associated cerebral hemorrhage. [32] A systematic review of studies that assessed the neuroprotective effects of minocycline in ischemic stroke showed that the antibiotic increases the viability of neurons and reduces the infarct volume after cerebral ischemia. [33]
Antiplatelet antibodies
Another antiadhesion monoclonal antibody strategy targets platelets. These antibodies inhibit platelet aggregation, potentially preventing additional ischemic injury during reperfusion, as well as promoting thrombolytic action. Such an antiplatelet drug, abciximab (ReoPro), was in phase III clinical stroke treatment trials, but an increased rate of symptomatic and fatal intracranial hemorrhage and lack of efficacy led to discontinuation of the trial after 808 patients were enrolled. [34] The rate of symptomatic or fatal hemorrhage was higher in a small cohort of patients that the study allowed to be treated within 3 hours of stroke that was present on awakening (13.6% abciximab vs 5% placebo, compared with 5.5% abciximab and 0.5% placebo in the primary cohort).
Citicoline
Citicoline is an exogenous form of cytidine-5'-diphosphocholine (CDP-choline) used in membrane biosynthesis. Citicoline may reduce ischemic injury by stabilizing membranes and decreasing free radical formation.
A phase II trial showed improved outcome in stroke patients treated with either a 500- or 2000-mg/d dose of citicoline. [35] A phase III trial then randomized patients in a 2:1 fashion to receive either 500 mg of citicoline or placebo orally in capsule form every day for 6 weeks. [36] Treatment began within 24 hours of symptom onset. There were 267 patients in the citicoline-treated group and 127 patients in the placebo group.
After 3 months, the citicoline-treated and placebo-treated groups showed no significant differences on selected assessments of functional outcome. There were no significant side effects. Despite similar mean baseline neurologic stroke scale scores, a higher percentage of patients treated with placebo had mild strokes. A post hoc subgroup analysis suggested that patients with more severe strokes (National Institutes of Health Stroke Scale >8) had better functional outcome with citicoline.
Another trial assessed the effect of citicoline on infarct size on magnetic resonance imaging (MRI) in patients with mild, moderate, and severe strokes. [37] Although this study also failed to show a significant difference between treated and untreated groups, there was a trend toward smaller infarct volumes in treated patients.
Another phase III trial was then performed, involving only moderate-to-large strokes. [38] The drug showed no adverse effects. Unfortunately, the company chose a novel endpoint (NIHSS score improvement >7 points), and the trial result was negative. If the company had instead used more conventional endpoints (eg, NIHSS = 0 or 1, Rankin = 0 or 1), the trial would have been positive.
The results of 4 clinical trials including 1372 patients (789 who received citicoline and 583 who received placebo) were pooled, [39] and the analysis showed that citicoline given within 24 hours from stroke symptom onset in patients with moderate-to-severe strokes was associated with a modestly increased probability of essentially complete recovery at 3 months. The results were assessed using a generalized estimating equation analysis to combine an NIHSS 1 or less, modified Rankin score of 1 or less, and a Barthel Index of 1 or less.
A large international trial, the International Citicoline Trial on acUte Stroke (ICTUS), enrolled 2298 patients with moderate-to-severe strokes within 24 hours of stroke onset. Unfortunately, this trial did not show efficacy for citicoline compared with placebo. [40] Citicoline has also been assessed in a pilot trial of 38 patients with intracerebral hemorrhage. [41] The drug was safe and showed trends toward efficacy.
A Cochrane review, which included 10 randomized, controlled trials involving 4281 participants, evaluated the benefits and harms of citicoline for the treatment of patients with acute ischemic stroke. Little to no difference was found between citicoline and its controls in all-cause mortality, disability or dependence in daily activities, severe adverse events, and functional recovery. [42]
Neuronal Healing
Fiblast, a basic fibroblast growth factor, could help regulate neuronal healing after ischemia. In a phase II safety trial, Fiblast was administered IV for up to 24 hours in acute stroke patients. [43] Although it was associated with transient leukocytosis, the drug otherwise appeared to be well tolerated and safe. A large trial was begun to evaluate the efficacy of Fiblast in stroke patients presenting within 6 hours of symptom onset. However, the trial was terminated because of poor risk-to-benefit ratios.
In an unblinded study of 12 stroke patients, autologous mesenchymal stem cells were delivered intravenously 36-133 days post-stroke. The study supported the feasibility and safety of delivery of a relatively large dose of autologous mesenchymal human stem cells into human subjects with stroke. Additional studies with stem cells in stroke patients are in progress. [44]
Yet another approach to promote brain repair after stroke is GSK249320, a monoclonal antibody that blocks myelin-associated glycoprotein, a molecule inhibiting axon outgrowth. A dosage-finding study showed that the drug was well tolerated but clinical trials in stroke are not currently in progress. [45]
Conclusions
The past few decades have produced a plethora of negative neuroprotective trial results. This is frustrating because all of these agents seemed effective in animal trials. There may be many reasons for this discrepancy. Perhaps the greatest difference between the animal and clinical studies is the timing of the drug treatment. The FAST-MAG trial was the first to use a time window that more closely approximated the treatment times used in the animal models but was also negative. Another potential explanation for the differences in outcomes is the relative youth and health of the animal models compared with that of the clinical population. Alternatively, it may be that the ischemic cascade is so complex in the human that targeting a single pathway may be ineffective.
A pattern of potential benefits in particular subgroups of patients, such as younger patients or those with moderate-severity strokes, has emerged in several trials. The pathophysiology underlying subgroup efficacy effects may be complex. Older patients tend to show poorer stroke outcomes both in treated and in placebo groups, which may narrow margins of efficacy. Patients with mild strokes may not show treatment effects, because the placebo group achieves high levels of spontaneous recovery. Mild strokes also are less likely to involve cortical structures and gray matter. If an agent has a primary effect on such highly metabolic gray-matter structures, it may not show efficacy in smaller strokes.
Ongoing studies will determine whether these treatments are indeed effective in their targeted subgroups. In agents hampered by side effects, such as certain of the NMDA receptor antagonists, modifications in structure may improve benefit-to-risk ratios. Finally, novel agents that have not yet entered clinical trials hold promise of potential efficacy.
In the future, optimal therapy may be achieved by combining neuroprotective agents with complementary mechanisms in a "stroke cocktail." Because these drugs will not necessarily display adverse effects in patients with hemorrhagic stroke, ambulance crews could begin administering this stroke cocktail in the field.
-
MIP image from a CTA demonstrates a filling defect or high-grade stenosis at the branching point of the right MCA trunk (red circle), suspicious for thrombus or embolus. CTA is highly accurate in detecting large vessel stenosis and occlusions, which account for approximately one-third of ischemic strokes.






