Practice Essentials
Sturge-Weber syndrome (SWS), also called encephalotrigeminal angiomatosis, is a neurocutaneous disorder with angiomas that involve the leptomeninges (leptomeningeal angiomas [LAs]) and the skin of the face, typically correlating to the ophthalmic (V1) and maxillary (V2) distributions of the trigeminal nerve. The hallmark of SWS is a facial cutaneous venous dilation, also referred to as a nevus flammeus or port-wine birthmark (PWB).
Signs and symptoms
SWS is generally diagnosed clinically, based on the typical cutaneous, central nervous system (CNS), and ocular abnormalities associated with it. Neurological signs include the following:
-
Developmental delay/intellectual disability
-
Learning problems
-
Attention deficit-hyperactivity disorder
Factors suggesting a progressive course of cortical damage in SWS include the following:
-
Initial focal seizures progressing to frequent, secondarily generalized seizures
-
Increasing seizure frequency and duration despite the use of antiepileptic drugs (AEDs)
-
Increasing duration of a transient postictal deficit
-
Increase in focal or diffuse atrophy - Determined by serial neuroimaging
-
Progressive increase in calcifications
-
Development of hemiparesis
-
Deterioration in cognitive functioning
Physical signs of SWS are as follows:
-
Port-wine birthmark
-
Macrocephaly
-
Ocular manifestations
-
Soft-tissue hypertrophy
-
Hemiparesis
-
Visual loss
-
Hemianopsia
Ocular involvement in SWS may include the following signs:
-
Hemangiomalike, superficial changes (which on histology demonstrate only venous dilation) in the eyelid
-
Buphthalmos
-
Glaucoma
-
Tomato-catsup color of the fundus (ipsilateral to the nevus flammeus) with glaucoma
-
Conjunctival and episcleral hemangiomas
-
Diffuse choroidal hemangiomas
-
Heterochromia of the irides
-
Tortuous retinal vessels with occasional arteriovenous communications
Ocular signs that may indicate the presence of infantile glaucoma include the following:
-
Corneal diameter of more than 12 mm during the first year of life
-
Corneal edema
-
Tears in the Descemet membrane (Haab striae)
-
Unilateral or bilateral myopic shift
-
Optic nerve cupping greater than 0.3
-
Any cup asymmetry associated with intraocular pressure (IOP) above the high teens
-
Optic nerve damage - Resulting in myopia, anisometropia, amblyopia, strabismus, and visual field defects
Diagnosis
In young patients, examination under anesthesia or deep sedation is necessary to confirm the diagnosis of glaucoma. Careful assessment in each eye of IOP, corneal diameter, cycloplegic refraction, axial length, and optic nerve cupping, as well as gonioscopic examination, is mandatory.
Cerebrospinal fluid (CSF) protein may be elevated, presumably secondary to microhemorrhage. Note that a major intracranial hemorrhage itself is rare in SWS, although microhemorrhage may be common.
Besides the clinical examination, the following have historically been the procedures of choice to establish the diagnosis of SWS: [1]
-
Skull radiography
-
Angiography
-
Computed tomography (CT) scanning
-
Magnetic resonance imaging (MRI)
-
MRI with gadolinium
-
Functional imaging - With single-photon emission computed tomography (SPECT) or positron emission tomography (PET) scanning
In the diagnosis of diffuse choroidal hemangioma, A-scan and B-scan ultrasonography may be useful diagnostic aids. B-scan ultrasonography characteristically shows a solid, echogenic mass, whereas A-scan ultrasonography demonstrates high internal reflectivity.
Electroencephalography (EEG) is used for the evaluation of seizures; it can also localize seizure activity when epilepsy surgery is considered for refractory seizures.
Management
Medical care in SWS includes anticonvulsants for seizure control, symptomatic and prophylactic therapy for headache, glaucoma treatment to reduce IOP, and laser therapy for the PWB.
Antiseizure medications
An antiseizure medication with efficacy in focal seizures is preferable in SWS. The chance of achieving seizure control with medical therapy in patients with SWS varies.
Glaucoma medications
The goal of treatment is control of IOP to prevent optic nerve injury. This can be achieved with the following agents:
-
Beta-antagonist eye drops - Decrease the production of aqueous fluid
-
Carbonic anhydrase inhibitors - Also decrease production of aqueous fluid
-
Adrenergic eye drops and miotic eye drops - Promote drainage of aqueous fluid
Dye laser photocoagulation
Treatment of the cutaneous PWB with dye laser photocoagulation has been helpful in reducing the cosmetic blemish from the cutaneous vascular dilatation. [2]
Surgery
Surgery is desirable in patients with SWS who have refractory seizures, glaucoma, or specific problems related to various SWS-associated disorders, such as scoliosis. [3]
Surgical procedures for seizures that are refractive to medical treatment include the following: [4]
-
Focal cortical resection
-
Hemispherectomy
-
Corpus callosotomy
-
Vagal nerve stimulation (VNS)
Procedures for the treatment of diffuse choroidal hemangiomas with retinal detachment include the following:
-
Cryotherapy and diathermy
-
Xenon arc or argon laser photocoagulation
-
Subretinal fluid drainage
-
Radiation therapy
A retrospective analysis of five patients treated for diffuse choroidal hemangioma associated with Sturge-Weber syndrome found that ruthenium-106 plaque radiotherapy is an effective and safe treatment. [5]
Surgical options for glaucoma in SWS include the following:
-
Goniotomy
-
Trabeculotomy
-
Full-thickness filtration surgery
-
Partial-thickness filtration surgery (trabeculectomy)
-
Combined trabeculotomy-trabeculectomy
-
Argon laser trabeculoplasty
-
Neodymium:yttrium-aluminum-garnet (Nd:YAG) laser goniotomy
-
Seton procedures
Background
Sturge-Weber syndrome (SWS), also called encephalotrigeminal angiomatosis, is a neurocutaneous disorder with angiomas that involve the leptomeninges (leptomeningeal angiomas [LAs]) and the skin of the face, typically in the ophthalmic (V1) and maxillary (V2) distributions of the trigeminal nerve. Extracranial angiomas and soft-tissue overgrowth also may occur in SWS. (See Pathophysiology, Etiology, and Clinical Presentation.)
Cutaneous manifestations
The hallmark of SWS is a facial cutaneous venous dilation, also referred to as a nevus flammeus or port-wine birthmark (PWB), which is present in as many as 96% of patients and is visible at birth (see the image below). The facial venous dilation appears as 1 or several dull red patches of irregular outline that are situated along, but are not limited to, the distribution of 1 or more divisions of the trigeminal nerve. [6, 7, 8] SWS belongs to a group of disorders collectively known as the phakomatoses ("mother-spot" diseases). (See Pathophysiology and Clinical Presentation.)
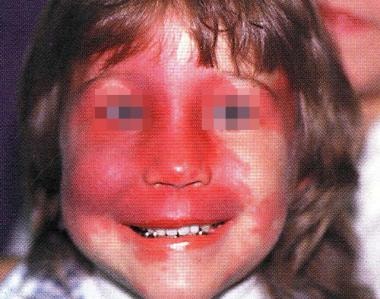
Laser therapy is available for the PWB. Although concerns have been raised that laser therapy to treat PWB might cause or worsen glaucoma or ocular hypertension; a 2009 retrospective review did not reveal evidence to support this. (See Treatment.) [2]
Neurological manifestations
In the brain, LAs demonstrated by structural neuroimaging may be unilateral or bilateral [9] ; unilateral angiomas are more common. Functional neuroimaging may demonstrate a greater area of involvement than structural neuroimaging. [10] This is called a structural-versus-functional mismatch. (See Workup.)
The neurological manifestations of SWS vary, depending on the location of the LAs (which most commonly are located in the parietal and occipital regions) and the secondary effects of the angiomas. These neurological morbidities include the following:
-
Seizures - May be intractable
-
Focal deficits - Including hemiparesis and hemianopsia, both of which may be transient (called "strokelike episodes")
-
Headaches
-
Developmental disorders - Including developmental delay, learning disorders, and intellectual disability; more common when angiomas are bilateral
Focal or generalized motor seizures usually begin in the first year of life. Profound seizure activity sometimes may be observed, with resultant further neurological and developmental deterioration. [11] Seizure control is thought to improve the neurological outcome, and epilepsy surgery may be beneficial for refractory seizures. Therefore, diagnosing and treating the disease early, before permanent damage to the brain occurs, is preferable. (See Prognosis and Treatment.)
Progressive, characteristic calcifications in the external layers of the cerebral cortex underlying the angiomatosis associated with ipsilateral cortical atrophy frequently develop and progress with age, occasionally extending anteriorly to the frontal and temporal lobes. (See Pathophysiology and Etiology.)
Certain CNS malformations have been associated with SWS; other neurocutaneous disorders are included in the differential diagnosis.
Ophthalmic manifestations
The primary complications involving the ipsilateral eye are buphthalmos and glaucoma, with treatment aimed at controlling the intraocular pressure (IOP) and preventing progressive visual loss and blindness.
Classification
SWS is referred to as complete when both CNS and facial angiomas are present, and incomplete when only the face or CNS is affected. The Roach Scale is used for classification, as follows: [12]
-
Type I - Facial and leptomeningeal angiomas; patient may have glaucoma
-
Type II - Facial angioma alone (no CNS involvement); patient may have glaucoma
-
Type III - Isolated LA (LA involvement only with no facial angioma); usually no glaucoma
Examples of ocular manifestations of SWS are shown in the images below.
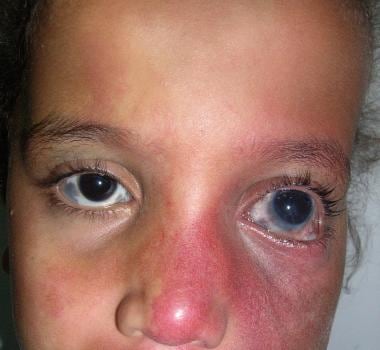 A child with Sturge-Weber syndrome that primarily affects the distribution of cranial nerve V2-3, with milder involvement of cranial nerve V1. Secondary glaucoma is evident. Ocular melanocytosis involving the sclera of both eyes is an associated finding. Image courtesy of Dr. Lamia Salah Elewa.
A child with Sturge-Weber syndrome that primarily affects the distribution of cranial nerve V2-3, with milder involvement of cranial nerve V1. Secondary glaucoma is evident. Ocular melanocytosis involving the sclera of both eyes is an associated finding. Image courtesy of Dr. Lamia Salah Elewa.
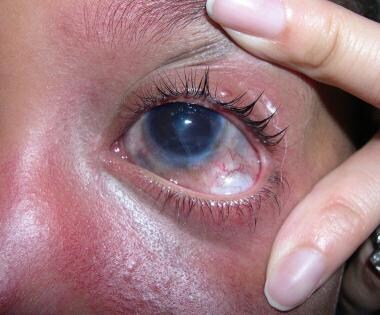 Close-up view of the left eye, showing the Ahmed valve implanted in the inferotemporal quadrant after multiple failed filtration procedures induced severe superior conjunctival scarring. Intraocular pressure (IOP) was controlled. Image courtesy of Dr. Lamia Salah Elewa.
Close-up view of the left eye, showing the Ahmed valve implanted in the inferotemporal quadrant after multiple failed filtration procedures induced severe superior conjunctival scarring. Intraocular pressure (IOP) was controlled. Image courtesy of Dr. Lamia Salah Elewa.
Pathophysiology
Sturge-Weber syndrome (SWS) is caused by residual embryonal blood vessels and their secondary effects on surrounding brain tissue. A vascular plexus develops around the cephalic portion of the neural tube, under ectoderm destined to become facial skin. Normally, this vascular plexus forms in the sixth week and regresses around the ninth week of gestation. Failure of this normal regression results in residual vascular tissue, which forms the angiomata of the leptomeninges, face, and ipsilateral eye. [13]
Neurologic dysfunction results from secondary effects on surrounding brain tissue, which include the following: [14] {ref15
-
Hypoxia
-
Ischemia
-
Venous occlusion
-
Thrombosis
-
Infarction
-
Vasomotor phenomenon
From a review of pathologic specimens, Norman and Schoene thought that blood flow abnormalities in the LA caused increased capillary permeability, stasis, and anoxia. [16] Garcia et al and Gomez and Bebin reported that venous occlusion may actually cause the initial neurologic event, either a seizure, transient hemiparesis, or both, thereby beginning the process. [17, 18]
A "vascular steal phenomenon" may develop around the angioma, resulting in cortical ischemia. Recurrent seizures, status epilepticus, intractable seizures, and recurrent vascular events may aggravate this steal further, with an increase in cortical ischemia, resulting in progressive calcification, gliosis, and atrophy, which in turn increase the chance of seizures and neurological deterioration. [19, 20]
Disease progression and neurological deterioration may occur in SWS. Although the actual LA is typically a static anatomic lesion, Maria et al, Reid et al, and Sujansky and Conradi have clearly documented the progressive nature of SWS. [21, 22, 23]
Udani et al followed the natural disease course and magnetic resonance imaging (MRI) findings of 9 patients with SWS. They found that earlier onset seizures correlated with more residual neurological deficits and worse focal cerebral atrophy and that in most cases the course stabilized after age 5 years. [24]
Seizure control, aspirin therapy, and early surgical treatment may prevent neurological deterioration. [1]
The main ocular manifestations (ie, buphthalmos, glaucoma) occur secondary to increased IOP with mechanical obstruction of the angle of the eye, elevated episcleral venous pressure, or increased secretion of aqueous fluid.
Etiology
The etiology of Sturge-Weber syndrome (SWS) is primarily likely associated with somatic mosaicism. Huq et al reported evidence of somatic mosaicism in 4 patients with SWS. [25, 26] Tissue samples were via skin biopsy from port-wine stains in 2 patients, and LAs from hemispherectomy in the other 2 patients. Inversion of chromosome arm 4q and trisomy 10 were seen in one patient each. Shirley et al identified a somatic activating c.548G->A mutation in GNAQ (on chromosome 9q21) in samples of affected tissues, in 23 out of 26 study participants with SWS. [27]
Malformed cortical vessels in SWS have been reported to be innervated only by noradrenergic sympathetic nerve fibers, [28] and increased endothelin-1 expression has also been seen in malformed intracranial vessels. These findings may suggest increased vasoconstriction in these abnormal blood vessels, as endothelin-1 is a peptide associated with vasoconstriction.
Fibronectin is a molecule important in regulating angiogenesis, maintenance of the blood-brain barrier, and blood vessel structure and function, as well as brain tissue responses to seizures. Comi et al reported that, in patients with SWS, decreased expression of fibronectin was noted in the leptomeningeal blood vessels, while increased expression was noted in the parenchymal vessels. The leptomeningeal blood vessel circumference was decreased, while blood vessel density was increased in SWS. [29]
Overall, in SWS, an activating somatic mutation in the GNAQ gene (p.Arg183Gln), seen in most cases [27] on chromosome 9 (at 9q21.2), appears to cause alterations in regulation of the structure and function of blood vessels, innervation of the blood vessels, and expression of extracellular matrix and vasoactive molecules.
Glaucoma
Glaucoma in SWS is produced by mechanical obstruction of the angle of the eye, elevated episcleral venous pressure, or hypersecretion of fluid by either the choroidal hemangioma or ciliary body. The anterior chamber angle abnormality is consistently seen in the infantile glaucoma cases in SWS, while increased episcleral venous pressure may have a key role in late-onset glaucoma cases in SWS. Decreased vision and blindness result from untreated glaucoma, with increased IOP leading to optic nerve damage. An acceptable range of IOP is 10-22 mm Hg. Enlargement of the eye occurs from the same mechanisms as glaucoma.
Epidemiology
Occurrence in the United States
According to the National Organization of Rare Disorders, Sturge-Weber Syndrome (SWS) occurs in one of every estimated 20,000 to 50,000 live births. [30] The inheritance is sporadic, and no regional differences in incidence have been identified. The incidences of the major clinical manifestations of SWS are listed in Table 1.
Table 1. Clinical Manifestations of Sturge-Weber Syndrome (Open Table in a new window)
Clinical Manifestation |
Incidence Rate |
Risk of SWS with facial PWB |
8% |
SWS without facial nevus |
13% |
Bilateral cerebral involvement |
15% |
Seizures |
72-93% |
Hemiparesis |
25-56% |
Hemianopia |
44% |
Headaches |
44-62% |
Developmental delay and intellectual disability |
50-75% |
Glaucoma |
30-71% |
Choroidal hemangioma |
40% |
International occurrence
SWS is found worldwide. Generally, the condition is easily diagnosed at birth or in early infancy based on the external clinical signs alone. However, the development of morbidity from secondary changes and complications occurs throughout life.
Age-related demographics
The typical SWS patient presents at birth with facial angiomas; however, not all children with facial angiomas and port-wine birthmark (PWB) have SWS, which raises certain diagnostic and prognostic concerns. [31]
In incomplete SWS (type III, Roach Scale), CNS angiomas occur without cutaneous features; therefore, no suspicion of SWS arises until a seizure or other neurologic problem develops. Thus, the diagnosis of SWS is not always straightforward.
Secondary glaucoma may present at any age, although early onset is the rule, with approximately 60% of glaucomas presenting at birth or in early infancy and another 30% presenting during childhood. The median ages reported for onset of visual symptoms related to secondary retinal changes range from age 8-20 years.
Prognosis
Neurologic and developmental morbidities in Sturge-Weber syndrome (SWS) include the following:
-
Seizures
-
Weakness
-
Strokes
-
Haeadaches
-
Hemianopsia
-
Intellectual disability
-
Developmental abnormalities
The development of seizures and their age of onset may correlate with the degree of neurological involvement. Neurological dysfunction increases with bilateral port-wine birthmark (PWB). Patients may experience complications related to refractory seizures and anticonvulsants, they may suffer visual loss and blindness from glaucoma, and they may display cosmetic deformities and other manifestations of soft-tissue involvement.
The glaucoma associated with SWS is a significant cause of morbidity because of its early onset and resistance to conventional forms of treatment. Glaucoma has been estimated to occur in 30-71% of patients with Sturge-Weber syndrome.
Prognostic factors
Factors predicting a poor outcome (or indicating potential need for surgery) in SWS include the following:
-
Early seizure onset
-
Extensive leptomeningeal angioma (LA)
-
Medically refractive seizures
-
Relapsing or permanent motor deficits
-
Headaches or mild trauma associated with transient motor deficits
-
Evidence of progressive neurologic damage
-
Focal seizures with subsequent generalization
-
Increasing seizure frequency and duration
-
Increasing duration of postictal deficits
-
Increasing focal or diffuse atrophy
-
Progressive atrophy or calcifications
-
Development of hemiparesis
-
Deterioration in cognitive functioning (loss of intellectual abilities)
Port-wine stain
Facial nevi are congenital macular lesions that can be progressive; they may be a light pink color initially and then progress to a dark red or purple nodular lesion. These may be isolated to the skin, associated with lesions in the choroidal vessels of the eye or the leptomeningeal vessels of the brain, or even located on other body areas. [32] A facial nevus, or PWB may be difficult to visualize in a patient with dark skin pigmentation.
Not all people with a PWB have SWS; the overall incidence of SWS has been reported to be 8-33% in individuals with a PWB. Several studies have evaluated this specifically.
A study by Enjolras et al indicated that in patients with a PWB, SWS occurs only when the nevus involves areas correlating to the V1 (ophthalmic) distribution of the trigeminal nerve. In their retrospective review, the investigators studied data from 106 patients with a facial PWB, 12 of whom had SWS and 4 of whom had glaucoma without pial lesions. No patients who had involvement of thea reas correlating to V2 (maxillary) and/or V3 (mandibular) distribution without V1 region involvement had SWS. [31]
Patients in the study who were considered to be at high risk for SWS were those with involvement of the entire V1 area; 11 of 25 patients with full V1 involvement had SWS. Patients with only partial involvement of V1 area were at low risk (only 1 of 17 patients had SWS).
In a study of 121 patients with facial nevi affecting the skin in the distribution of the trigeminal nerve, Bioxeda et al concluded that only those persons with V1 region involvement were at risk for epilepsy or glaucoma. The investigators found that glaucoma and epilepsy were present in 23 (17%) and 17 (14%) patients, respectively, with V1 region involvement occurring in all 40 of these individuals. [33]
The investigators also found that the facial nevi were located predominantly over the region correlating to the distribution of V2 branch of the trigeminal nerve in 88% of these individuals, either isolated to the V2 branch region or also involving the V1 and/or V3 branch regions. An extrafacial PWB was more common when V3 region was involved. The lesions were unilateral in 86% of patients, and bilateral in 14% of them.
In a similar, but larger, study, Tallman et al found that only patients with a PWB involving the distributions of the V1 and V2 branches of the trigeminal nerve had CNS or eye involvement. The investigators reported on 310 patients with facial nevi, 85% of who had unilateral involvement, 15% of whom had bilateral involvement, and 68% of who had involvement of more than 1 dermatome. Overall, in patients with trigeminal involvement, only 8% had CNS and eye involvement; 24% of those with bilateral lesions had eye or CNS involvement, compared with only 6% of patients with unilateral lesions. [34]
Tallman and colleagues also found that all patients with eye or CNS involvement had lesions on the eyelids; 91% of these had both upper and lower eyelid involvement, whereas 9% had only lower eyelid involvement. No patients with upper eyelid involvement alone had eye or CNS involvement. Three of 16 patients with involvement of V1, V2, and V3 regions had eye and/or CNS involvement. The authors recommended screening for glaucoma and CNS involvement when the PWB involved the eyelids, with unilateral V1, V2, and V3 region lesions, or with bilateral lesions.
Patients identified by the Sturge-Weber Foundation had a different pattern of involvement—170 of 171 patients had a craniofacial PWB, with unilateral involvement in 83 patients (49%) and bilateral involvement in 86 patients (51%).
Note that an extrafacial PWB may have associated intracranial abnormalities; for example, in Klippel-Trenaunay-Weber syndrome, neuroimaging may show findings similar to those of SWS, [35] and a cervical PWB has been associated with occipital calcifications. [32]
Seizures
The incidence of epilepsy in patients with SWS is 75-90%. Seizures result from cortical irritability caused by cerebral angioma, through mechanisms of hypoxia, ischemia, and gliosis. Dual pathology, such as microgyria, also may be present, which also contributes to epileptogenesis. Seizures may be intractable in some patients.
Garcia et al reported that a child with SWS could have an early normal neurologic course, with a seizure as the presenting manifestation of a neurologic problem. [17]
In a survey of cases identified by the Sturge-Weber Foundation, [8] seizures occurred in 136 of 171 patients, with the age of onset ranging from birth to 23 years and the median age of onset being 6 months. About 75% of the patients had onset during the first year of life; 86%, before age 2 years; and 95%, before age 5 years. Seizures occurred in 71% of those with unilateral and 87% of those with bilateral disease.
Bebin and Gomez, from the Mayo Clinic, reported that seizures occurred in 80% of patients with SWS (72% with unilateral involvement vs 93% with bilateral involvement), with the median age of onset being 8.5 months in patients with unilateral PWB and 4 months in patients with bilateral PWB. [36]
Oakes reported seizures in 24 (80%) of 30 patients with SWS, with a mean age of onset of 6 months. [37]
Bebin and Gomez reported an earlier onset of seizures in patients with bilateral involvement (mean ages of seizure onset were 6 months with bilateral disease and 24 months with unilateral disease). [36] Pascual-Castroviejo et al showed that patients with more frequent seizures tended to have an earlier seizure onset (mean seizure onset at age 5-6 months compared with a mean onset at age 2 years in those with less severe involvement). [38]
Pascual-Castroviejo et al reported seizures in 32 (80%) of 40 patients. Seizures began during a febrile illness in 10 patients (31%; fever could be a precipitant at any age), while infantile spasms occurred in 2 patients (6%). [38]
Developmental delay
In the patients reported by the Sturge-Weber Foundation, 50% had complete control and 39% had partial control of seizures with medications. Those with a later age of seizure onset had a lower incidence of developmental delay and fewer special educational needs.
The onset of seizures prior to age 2 may suggest a greater chance of refractory epilepsy and intellectual disability. [1] Patients with refractory seizures are more likely to have intellectual disability, since those individuals have more extensive brain involvement.
In a report by Sujansky and Conradi, using data obtained through the Sturge-Weber Foundation, [23] overall developmental delay occurred in 97 (58%) of 168 patients with SWS. Early developmental delay, however, occurred in 71% of patients with seizures and in only 6% of those without seizures. Patients with a later seizure onset also had a lower incidence of developmental delay and fewer special education needs.
However, even with seizure onset within the first year, Erba and Cavazzuti reported satisfactory control in 50% of patients, with 30% of patients being seizure free for at least 2 years. Among the other patients, 17% had an average of 1 seizure per month, and 33% were considered to have poorly controlled seizures, defined as greater than 1 seizure per week. [39] Therefore, early seizures may not predict either the severity of subsequent epilepsy or severe intellectual disability.
Maria et al divided their patients into 2 groups by age for a longitudinal study—those aged 1-3 years versus those aged 10-22 years—and found no difference in clinical outcomes with early onset seizures. [21]
Focal versus generalized seizures
Since the lesion responsible for epilepsy in SWS is focal, the majority of seizures are focal seizures. In a study of 76 patients, Bebin and Gomez reported partial seizures in 35 patients (46%), generalized seizures in 15 of them (20%), and both in 26 of them (34%). [36]
Pascual-Castroviejo et al reported seizures in 32 (80%) of 40 patients with SWS; 22 (69%) of them had focal seizures contralateral to the PWB, with subsequent generalization in 6 patients; 8 patients (25%) had generalized seizures at onset, and infantile spasms occurred in 2 patients (6%). [38]
Status epilepticus
Prolonged seizures cause neurologic injury secondary to metabolic disturbances such as hypoxemia, hypoglycemia, hypotension, ischemia, and hyperthermia.
In an already compromised vascular system, such as a vascular steal from the angioma, seizures are more likely to cause injury, even when short. Episodes of status epilepticus are, therefore, especially dangerous in SWS. [40]
Strokelike episodes
Transient episodes are referred to as strokelike episodes. These occurred in 14 (70%) of 20 patients described by Maria et al. [21] Garcia et al reported recurrent thrombotic episodes. [17] Stroke also may occur. The incidence of neurologic deficit is higher in adults; Sujansky and Conradi reported an occurrence in 34 (65%) of 52 patients, [41] results that demonstrated the progressive nature of SWS.
Hemiparesis
The incidence of hemiparesis is approximately 33% in patients with SWS, varying from 25-56%. The disorder occurs secondary to ischemia with venous occlusion and thrombosis. Commonly, transient weakness may occur with seizures and may increase with recurrent seizures. Transient hemiplegia may be accompanied by migraine headache, suggesting a vascular mechanism. However, Jung et al reported a case of a woman with headache and left hemiparesis but no neuroimaging evidence of acute thrombosis formation or recent vascular event. [42]
Hemianopsia
The mechanism for hemianopsia is similar to that for hemiparesis and is dependent on the location of the lesions. Uram and Zullabigo reported hemianopsia in 11 (44%) of 25 patients. [43] In 2009, Shimakawa et al reported a rare case of recurrent homonymous hemianopsia that became permanent. [44]
Developmental delay and intellectual disability
Related to the degree of neurologic involvement, developmental delay and intellectual disability occur in 50-60% of patients with SWS; they are more likely to exist in patients with bilateral involvement. [9] Bebin and Gomez reported normal mental functioning in only 8% of patients with bilateral SWS. [36]
In a detailed study of 10 patients with SWS, Maria et al found developmental delay and learning problems in all 10 and attention deficit hyperactivity disorder in 3. [45] Using a combination of computed tomography (CT) scanning, MRI, and functional imaging with single-photon emission CT (SPECT) scanning, abnormalities were found bilaterally the majority of patients, including extensive abnormalities in glucose metabolism and cerebral perfusion. These results may account for the high incidence of developmental delay, intellectual disability, and learning problems seen in SWS.
Seizures are also associated with a higher incidence of intellectual disability, and regression may be related to the frequency and severity of seizures as well.
Alkonyi et al reported on 14 patients with bilateral SWS who had an asymmetrical pattern on positron-emission tomography (PET) scanning and found that bilateral frontal and temporal hypometabolism was associated with poor developmental outcome. Good seizure control and only mild to moderate developmental impairment was seen in about 50% of the patients with bilateral SWS. [46]
Headaches
Occurring secondary to vascular disease, these have the symptoms of a migraine headache and are considered "symptomatic migraines."
In the aforementioned study by the Sturge-Weber Foundation, headaches occurred in 132 (77%) of 171 of patients of all ages and in 28 (62%) of 45 adults. In reports by Maria et al, headaches occurred in 60% of patients. [21, 45, 1]
In a specific study of headaches in SWS reported by Klapper, 71 patients identified by the Sturge-Weber Foundation responded to a questionnaire about headaches. [47] Migraine headache occurred in 28% of patients, and neurologic deficits occurred in 58% of these patients during the migraine. The prevalence of migraine was 31% in children younger than 10 years, much greater than the 5% prevalence in the general population.
Ocular manifestations
Glaucoma typically occurs in SWS only when the PWB involves the eyelids. The incidence ranges from 30-71%. Glaucoma may be present at birth but can develop at any age, even in adults. [48, 49]
Treatment includes yearly examinations to look for optic nerve damage (with measurement of IOP and visual fields) and for corneal diameter and refractive changes in children.
Glaucoma usually occurs only with an ipsilateral facial PWB, although the glaucoma may be bilateral when facial involvement is bilateral. Contralateral glaucoma may develop, although rarely. Glaucoma also may occur without neurologic involvement (Type II, Roach Scale).
Sullivan et al reviewed ocular abnormalities in 51 patients with SWS. [50] Of these, 36 patients (71%) had glaucoma, with onset before age 24 months in 26 (51%) patients; 35 (69%) patients had conjunctival or episcleral hemangiomas; and 28 (55%) patients had choroidal hemangiomas.
With time, choroidal hemangioma may cause other secondary changes, including the following:
-
Retinal pigment epithelium degeneration
-
Fibrous metaplasia
-
Cystic retinal degeneration
-
Retinal detachment
Retinal vascular tortuosity, iris heterochromia, optic disc coloboma, and cataracts have also been seen in patients with SWS.
The Sturge-Weber Foundation data show that 82 (48%) of 171 patients had glaucoma. Of these patients, 61% developed glaucoma during the first year of life; a second peak occurred in children aged 5-9 years, when it developed in another 11 (15%) patients.
Endocrine problems
An increased rate of growth hormone deficiency was found in SWS, as identified from a registry of 1653 patients. [51] Comi et al found central hypothyroidism in 2 children out of 83 (2.4%) with SWS and brain involvement, indicating that central hypothyroidism is much more prevalent in such patients than it is in the general population. [52]
Additional morbidities
The Sturge-Weber Foundation survey indicated that other abnormalities occurred in all 171 patients in the study. These included other cutaneous lesions in all patients and body asymmetry in 164 (96%) of 171 patients, with soft-tissue hypertrophy occurring in 38 (23%) of the 164 patients and scoliosis existing in 11 (6.7%) of the 164 patients. Basal cell carcinoma has been reported to occur within a PWB. [53]
Adults with SWS
Few data are available on adults with SWS. Sujansky and Conradi studied the outcomes in 52 adults older than 18 years who had SWS and were identified by the Sturge-Weber Foundation. [41] The age of onset of glaucoma ranged from 0-41 years, with a median of 5 years.
Seizures occurred in 83% of the patients, glaucoma in 60%, and a neurologic deficit—such as stroke, paralysis, spasticity, or weakness—in 65%. The age of onset of seizures ranged from 0-23 years, with a median of 6 months.
Seizure outcome was known in 41 patients in the study, with full control attained in 11 (27%) patients, a decrease in seizures achieved in (49%) 20 patients, and no improvement found in 10 (24%) patients. The morbid conditions associated with these seizures are listed in Table 2, below.
Headache occurred in 28 (62%) of 45 patients, with age of onset ranging from early childhood to age 38 years and with a median age of onset of 18 years. The headache frequency could be determined in 23 patients: daily in 9 patients (39%), 1-4 times per week in 4 (17%), 1-2 times per month in 6 (26%) patients, and rare in 4 patients (17%).
Headaches were associated with increased discoloration of facial PWB, auras, nausea/vomiting, dysarthria, dizziness, and feelings of facial pulsation.
Table 2. Developmental Morbidity Associated with Seizures in Adults with SWS (Open Table in a new window)
|
With Seizures (%) |
Without Seizures (%) |
Developmental delay |
45 |
0 |
Emotional/behavioral problems |
85 |
58 |
Need for special education |
71 |
0 |
Employability |
46 |
78 |
Patient Education
Counseling may assist with treatment. [54] Support groups for persons with Sturge-Weber syndrome include The Sturge-Weber Foundation, PO Box 418, Mount Freedom, NJ, 07970-0418.
-
A child with Sturge-Weber syndrome with bilateral facial port-wine stain.
-
Cranial CT scan showing calcifications.
-
MRI image in Sturge-Weber syndrome.
-
Single-photon emission computed tomographic scan in Sturge-Weber syndrome.
-
A child with Sturge-Weber syndrome that primarily affects the distribution of cranial nerve V2-3, with milder involvement of cranial nerve V1. Secondary glaucoma is evident. Ocular melanocytosis involving the sclera of both eyes is an associated finding. Image courtesy of Dr. Lamia Salah Elewa.
-
Close-up view of the left eye, showing the Ahmed valve implanted in the inferotemporal quadrant after multiple failed filtration procedures induced severe superior conjunctival scarring. Intraocular pressure (IOP) was controlled. Image courtesy of Dr. Lamia Salah Elewa.
-
T1-weighted, axial magnetic resonance imaging (MRI) scans demonstrate left cerebral hemiatrophy associated with leptomeningeal angiomatosis. Image courtesy of Dr. Lamia Salah Elewa.
-
Ocular ultrasonogram of the posterior segment demonstrating the diffuse choroidal thickening seen in a diffuse choroidal hemangioma with "tomato-catsup fundus." Image courtesy of Dr. Lamia Salah Elewa.
-
Choroidal hemangioma. Image courtesy of Thomas M. Aaberg, Jr, MD.
-
Choroidal hemangioma. Image courtesy of Thomas M. Aaberg, Jr, MD.
-
Circumscribed hemangioma. Image courtesy of F. Ryan Prall, MD.
-
Circumscribed hemangioma. Image courtesy of F. Ryan Prall, MD.
-
B-scan of a choroidal hemangioma showing medium to high internal reflectivity. This is a circumscribed choroidal hemangioma. The patient was not diagnosed with Sturge-Weber Syndrome. Image courtesy of Abdhish R Bhavsar, MD.
Tables
- Table 1. Clinical Manifestations of Sturge-Weber Syndrome
- Table 2. Developmental Morbidity Associated with Seizures in Adults with SWS
- Table 3. Summary of Work-up Findings in Sturge-Weber Syndrome
- Table 4. Seizure Control in Sturge-Weber Syndrome
- Table 5. Surgical Results of Hemispherectomy and Limited Resection from 3 Centers
Clinical Manifestation |
Incidence Rate |
Risk of SWS with facial PWB |
8% |
SWS without facial nevus |
13% |
Bilateral cerebral involvement |
15% |
Seizures |
72-93% |
Hemiparesis |
25-56% |
Hemianopia |
44% |
Headaches |
44-62% |
Developmental delay and intellectual disability |
50-75% |
Glaucoma |
30-71% |
Choroidal hemangioma |
40% |
|
With Seizures (%) |
Without Seizures (%) |
Developmental delay |
45 |
0 |
Emotional/behavioral problems |
85 |
58 |
Need for special education |
71 |
0 |
Employability |
46 |
78 |
Procedure |
Findings |
CSF analysis |
Elevated protein |
Skull radiography |
Tram-track calcifications |
Angiography |
Lack of superficial cortical veins Non-filling dural sinuses Abnormal, tortuous vessels |
CT scanning |
Calcifications, tram-track calcifications Cortical atrophy Abnormal draining veins Enlarged choroid plexus Blood-brain barrier breakdown (during seizures) Contrast enhancement |
MRI |
Gadolinium enhancement of leptomeningeal angiomas (LAs) Enlarged choroid plexus Sinovenous occlusion Cortical atrophy Accelerated myelination |
SPECT scanning |
Hyperperfusion, early Hypoperfusion, late |
PET scanning |
Hypometabolism |
Electroencephalography (EEG) |
Reduced background activity Polymorphic delta activity Epileptiform features |
Study |
Complete |
Partial |
Refractory/No Control |
Gilly et al [97] |
NA* |
NA |
37% |
Sujanski and Conradi [41] (adults) |
27% |
49% |
24% |
50% |
39% |
11% |
|
Pascual-Castroviejo et al [38] |
47% |
12% |
28% |
Oakes [37] |
10% |
NA |
83% |
Sassower et al [89] |
NA |
NA |
43% |
Arzimanoglou and Aicardi [96] |
NA |
NA |
39% |
Erba and Cavazzuti [39] |
50% |
NA |
NA |
NA |
NA |
32% |
|
*NA = not available |
|||
Center |
Hemispherectomy |
Seizure Free |
Limited resection |
Seizure Free |
Improved |
Toronto |
12 |
11 |
11 |
8 |
2 |
Paris |
5 |
5 |
15 |
7 |
8 |
Boston |
9 |
8 |
6 |
3 |
0 |
Total |
26 |
24 |
32 |
18 |
10 |
24 of 26 patients with hemispherectomy - Seizure free 28 of 32 patients with limited resection - Seizure free or improved |
|||||

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Approach Considerations
- Pharmacologic Treatment of Seizures
- Pharmacologic Treatment of Glaucoma
- Headaches
- Strokelike Events
- Port-Wine Birthmark
- Surgical Treatment of Seizures
- Hemispherectomy
- Surgical Treatment of Diffuse Choroidal Hemangiomas
- Glaucoma Surgery
- Correction of Anisometropia and Strabismus
- Consultations
- Long-Term Monitoring
- Show All
- Guidelines
- Medication
- Questions & Answers
- Media Gallery
- Tables
- References




