Practice Essentials
Progressive supranuclear palsy (PSP) is a neurodegenerative disease (see the image below) whose characteristics include supranuclear, initially vertical, gaze dysfunction accompanied by extrapyramidal symptoms and cognitive dysfunction. The disease usually develops after the sixth decade of life, and the diagnosis is purely clinical.
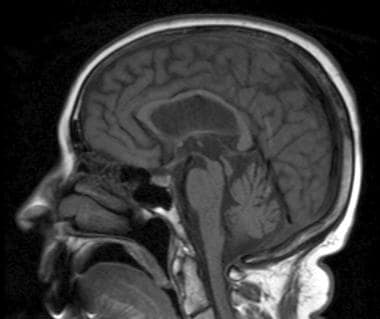 Sagittal T1-weighted image shows atrophy of midbrain, preservation of pontine volume, and atrophy of the tectum, suggestive of progressive supranuclear palsy (Steele-Olszewski-Richardson disease).
Sagittal T1-weighted image shows atrophy of midbrain, preservation of pontine volume, and atrophy of the tectum, suggestive of progressive supranuclear palsy (Steele-Olszewski-Richardson disease).
Signs and symptoms
The onset of PSP is insidious and usually includes a prolonged phase marked by the following symptoms:
-
Fatigue
-
Headaches
-
Arthralgias
-
Dizziness
-
Depression
Patients also experience subtle personality changes, memory problems, and pseudobulbar symptoms, which are often more evident to the family than to the patient. The initial symptoms can often involve unexplained imbalance or falls.
The cardinal manifestations of PSP are as follows:
-
Supranuclear ophthalmoplegia
-
Pseudobulbar palsy
-
Prominent neck dystonia
-
Parkinsonism
-
Behavioral, cognitive, and gait disturbances that cause imbalance
-
Frequent falls/impaired postural reflexes
Findings on physical examination can include the following:
-
Poor postural reflexes, axial rigidity greater than appendicular rigidity, and dysarthria (monotone with slight hypophonic quality)
-
Absence of cogwheeling or tremor
-
Widely based and unstable gait
-
Bradykinesia with masked facies and a startled expression
-
Retrocollis may be present
Visual signs and symptoms
-
Slow vertical saccades and square wave jerks on ocular examination (early signs)
-
Supranuclear ophthalmoplegia (classic gaze palsy in PSP)
-
Downgaze typically involved before upgaze
-
Improvement in supranuclear vertical gaze limitation after extravolitional pathway activation with the vestibular ocular reflex (VOR) or the Bell phenomenon
-
Nearly continuous square wave jerks commonly observed with fixation
-
Impairment of convergence eye movements (may cause diplopia)
-
Eyelid retraction, eyelid opening or closing apraxia, blepharospasm, or lid lag
-
Complete ophthalmoparesis in advanced PSP
Cognitive symptoms
-
Slowed cognitive processing, sequencing and planning difficulties, mild memory difficulty, and apathy (generally more prominent in late disease)
-
High apathy scores coupled with low agitation and anxiety scale scores on Neuropsychiatric Inventory testing
See Clinical Presentation for more detail.
Diagnosis
The diagnosis of PSP is clinical. Key features typically develop over time. Participants in a National Institute of Neurological Disorders and Stroke (NINDS)/Society for PSP conference have formulated and validated clinical research criteria for the diagnosis of PSP. [1] In this system, criteria for possible PSP are as follows:
-
Gradually progressive disorder with onset at age 40 years or older
-
Either vertical supranuclear palsy or both slowing of vertical saccades and prominent postural instability with falls in the first year of onset
-
No evidence of other diseases that can explain the clinical features
Criteria for probable PSP are vertical supranuclear palsy with prominent postural instability, falls in the first year of onset, and other features of possible PSP, as follows:
-
Abnormal neck posture, especially retrocollis
-
Poor or absent response of parkinsonism to levodopa therapy
-
Early dysphagia and dysarthria
-
Early cognitive impairment with at least 2 of the following: apathy, abstract thought impairment, decreased verbal fluency, imitation behavior, or frontal release signs
Criteria for definite PSP are as follows:
-
History of probable or possible PSP
-
Histopathologic evidence that is typical of the disease
The workup in patients with suspected PSP is directed principally at eliminating other diagnoses (eg, Whipple PCR to eliminate possible Whipple disease). MRI offers little help in the early stages of PSP, but may reveal the following abnormalities in some advanced cases [2, 3, 4, 5, 6] :
-
Atrophy of the midbrain (see the image below) with cisternal and ventricular dilatation
-
Thinning of the quadrigeminal plate
-
Dilation of the third ventricle
-
A nonspecific finding of increase in proton density images in the periaqueductal gray matter, compatible with gliotic changes
Sleep studies in patients with PSP show the following abnormalities, although these are not specific for PSP:
-
Diminished total sleep time
-
Increased awakenings
See Workup for more detail.
Management
No medication is effective in halting the progression of PSP; however, medications that may provide modest symptomatic improvement include the following:
-
Dopamine agonists
-
Tricyclic antidepressants
-
Methysergide
-
OnabotulinumtoxinA: May be useful for rigidity (nuchal rigidity in particular) and dystonia (eg, blepharospasm, bruxism, and focal limb dystonia) [11]
-
Methylcellulose or methyl alcohol eyedrops: For relief of chronic conjunctivitis from reduced blink rate
Physical therapy and rehabilitation medicine involvement may help maximize ambulation safety and facilitate instruction in the use of a walker, wheelchair, or other aids.
See Treatment and Medication for more detail.
Background
Progressive supranuclear palsy (PSP), also known as Steele-Richardson-Olszewski syndrome, is a neurodegenerative disease that affects cognition, eye movements, and posture. [12, 13] PSP was first described as a clinicopathologic entity in 1964. Characteristics include supranuclear, initially vertical, gaze dysfunction accompanied by extrapyramidal symptoms and cognitive dysfunction. The disease usually develops after the sixth decade of life, and the diagnosis is purely clinical. Currently, no therapy is proven to be effective.
Pathophysiology
Pathologically, PSP is defined by the accumulation of neurofibrillary tangles in the brain. [14] Different rates, localizations, and patterns of the accumulation of phosphorylated tau protein may account for the variation in clinical phenomena seen in patients with PSP.
The tau protein
The tau protein is important in maintaining neuronal morphology through microtubule binding. Abnormalities of this protein have been noted in several neurodegenerative diseases. Under abnormal circumstances, the normally soluble tau protein may collect in insoluble protease-resistant helical filaments. The exact triggers for the conversion from normal tau to the aggregate form are not completely understood.
This model shares some characteristics with prion disease (Creutzfeldt-Jakob disease), in which an abnormal insoluble prion protein isoform accumulates.
Work by Conrad et al demonstrated the overrepresentation of the homozygous tau A0 allele in patients with PSP compared to controls. [15] Accordingly, the tau A0 allele may be a genetic marker of increased susceptibility to the PSP pathophysiology. However, the tau A0 allele status is not required or sufficient to predict the occurrence of PSP.
Although the e4 allele of the apoprotein E gene (ApoE) is a significant risk factor for the development of Alzheimer disease and is overrepresented in individuals with Lewy body disease, it is not associated with PSP, Parkinson disease, or alcoholic dementia.
Liao et al found that during near viewing, the translational vestibulo-ocular reflex responses in patients with PSP were, on average, only 12% of those of control subjects, which suggested that abnormal otolith-mediated reflexes may be at least partly responsible for the frequent falls in patients with PSP. [16] The amplitude of vestibular-evoked myogenic potentials was also significantly lower in PSP patients than in normal control subjects.
Etiology
The cause of PSP remains unknown. Most cases appear to be sporadic. Both environmental and genetic influences have been postulated. To date, there have been only a few epidemiologic studies investigating PSP associations.
In a questionnaire survey carried out on a cohort of 75 patients with PSP and matched controls, Golbe et al obtained information on exposures (eg, to hydrocarbons, pesticides and herbicides), urban versus rural living, occupation, trauma, education level, maternal age, and family history of neurologic diseases; patients with PSP were less likely than controls to have completed 12 years of education. [17] The authors speculate that education level may be a marker for more direct risk factors (eg, early life nutrition or occupational or residential exposure).
The role of heredity in the pathophysiology of PSP remains elusive. Although there are anecdotal reports in the literature that describe apparent familial PSP, several larger series have not noted this association. In one case-control questionnaire, a trend toward relatives with parkinsonism was reported.
Tetrud et al reported the occurrence of autopsy-proven PSP in a brother-sister pair. [18] Both developed parkinsonism in the eighth decade of life and subsequently exhibited typical features of PSP over the next 5 years. Their mother and possibly their maternal grandfather experienced a parkinsonian syndrome, and essential tremor was noted in their father and 2 of the brother’s 3 children. The probands exhibited typical pathologic features of PSP upon autopsy.
Although the current absence of a large kindred with PSP precludes molecular linkage studies, the authors suggest that pairs such as those in their report could be pooled for analysis [18] ; such occurrences are quite rare.
Kaat et al reported that of 57 (33%) of 172 patients with PSP had at least 1 first-degree relative who had dementia or parkinsonism, compared with 25% of control subjects. [19] More first-degree relatives with parkinsonism were observed in PSP patients than in controls.
Although most cases of PSP appear to be sporadic, rare genetically determined forms may exist. Garcia de Yebenes et al studied a 5-generation family in which PSP was transmitted as an autosomal dominant trait, [20] finding 2 instances of male-to-male transmission. The proband had the classic presentation of this disorder beginning with axial rigidity, slowness of movement, and gait difficulty. Over a period of 2 years, he progressed to complete vertical gaze palsy, axial dystonia, retrocollis, and generalized severe akinesia.
Postmortem examination demonstrated neurofibrillary tangles (NFTs) and gliosis without prominent senile plaques, the same pathology that was observed in the sporadic cases of PSP described by Steele et al. [21]
In addition, Garcia de Yebenes et al described 6 other families with multiple affected individuals. These included 2 in which a parent was affected, suggesting autosomal dominant inheritance, and 1 family in which parental consanguinity occurred, suggesting recessive inheritance. [20] In the cohort studied by Kaat et al, 12 of 172 patients with PSP (7%) fulfilled the criteria for an autosomal dominant mode of transmission. [19]
Epidemiology
United States
A population-based study in New Jersey by Golbe et al revealed an overall prevalence of 1.39 cases per 100,000 population. The male prevalence was 1.53 in 100,000, whereas the female prevalence was 1.23 in 100,000; this finding was in accordance with a previously noted slight male preponderance. The adjusted prevalence ratio among patients older than 55 years was 7 in 100,000. [22] Prevalence data derived from tertiary centers suggest that PSP affects 4-6% of patients with parkinsonism. [23]
International
The incidence of PSP has been assessed in Perth, Australia; crude incidence rates are 3-4 per million cases per year, approximately 5% of the incidence of Parkinson disease. [24]
Age-, sex-, and race-related demographics
The mean age at onset is approximately 63 years (range, 44-75 years). [22, 25] The median interval between onset and diagnosis is 3 years (range, 0.5-9 years). PSP has a slight male predominance in most studies. According to Kristensen, the male-to-female ratio is 1.5:1. [26]
Most reported cases have been in whites. The affected cohort in Golbe’s study consisted entirely of white persons; however, the survey population included only 5.7% black individuals, thus preventing any meaningful analysis regarding race. [22]
Prognosis
Studies of cohort patients dying under surveillance suggest that PSP is usually fatal within approximately 6 years of onset (range, 2-17 years); life table analysis among the entire cohort of Golbe et al revealed a median disease duration of 9.7 years. [22] Conflicting reports exist regarding the influence of age at diagnosis on survival; Golbe found a tendency for younger patients to survive longer, although this is not a uniform finding among other studies. [22, 25]
The primary causes of death in patients with PSP are infections and pulmonary complications (eg, pneumonia) that are frequently related to immobility. Often, the primary morbidity relates to imbalance leading to immobility, though dementia, visual symptoms, and dysphagia are major concerns. About 50% of patients require some aid to walk within 3 years of the initial onset of symptoms. The usual interval from initial symptom occurrence to the need for a cane or a walker is 3.1 years, and the interval to confinement to a chair or bed is 8.2 years. [22]
Patient Education
The primary educational efforts related to PSP are focused on the patient and family and aimed at fostering an understanding of the disease, the patient’s prognosis, potential complications, and effective coping mechanisms. Many patients and families benefit from contact with and participation in a support group. Further information is available from CurePSP and The Parkinson’s Institute.
For patient education resources, see the Dementia Center, as well as Progressive Supranuclear Palsy.
-
Sagittal T1-weighted image shows atrophy of midbrain, preservation of pontine volume, and atrophy of the tectum, suggestive of progressive supranuclear palsy (Steele-Olszewski-Richardson disease).
-
Characteristic facial appearance of patient with progressive supranuclear palsy.






