Background
The biopsy of skeletal muscle tissue, typically termed muscle biopsy, plays an integral role in evaluation of the patient with neuromuscular disease. With some exceptions, it is an essential element in the assessment of a patient with suspected myopathy. [1, 2, 3] It is also sometimes indicated for the diagnosis of various systemic disorders that can be accompanied by pathology of skeletal muscle tissue.
In addition, muscle biopsy is relatively commonly part of the evaluation of suspected neuropathic disease, sometimes for the purpose of assisting the clinician to distinguish a neurogenic disorder with an atypical presentation from a primary myopathic disorder and in other cases for the purpose of increasing the diagnostic yield when there is a clinical concern that vasculitis might be present.
The surgical procedure to obtain a muscle biopsy is relatively simple and poses little risk to the patient. It is, however, a specialized procedure and must be performed properly in order to maximize the information it can yield for the benefit of the patient.
The clinician must first arrive at a rational differential diagnosis by synthesizing information obtained from the clinical history, physical examination, and laboratory and electrodiagnostic studies. This information is used to influence the details of the muscle biopsy procedure. The choices of the right time for biopsy, which muscle to select, how many specimens to obtain, and how to handle the specimen immediately following excision are individualized for each patient on the basis of these clinical features.
After the biopsy specimen arrives in the pathology laboratory, it undergoes multiple procedures and studies. The pathologist uses knowledge of the clinical features for guidance in interpretation of the constellation of pathologic findings in the biopsy, as well as to help determine whether additional studies are indicated for a given biopsy on the basis of the composite of the specific clinical features and biopsy findings.
Muscle biopsy, therefore, is somewhat complex, in that an optimal outcome requires coordination of the clinician, surgical team, pathologist, and technical staff in the pathology laboratory. Because muscle biopsies are often processed and interpreted at specialized centers, it may also be necessary to involve a courier service or shipping company in the process.
A portion of this article serves as a primer on the technical aspects of muscle biopsy, which are critical for the success of the procedure. The aim is to help familiarize the reader with the basic concepts of how biopsies should be performed and what happens to the tissue that is obtained, as well as to provide some background information to help nonpathologists understand the pathology reports that they receive.
An introduction to the clinical features of neuromuscular disease is included in this article (see Clinical Features of Neuromuscular Disorders, below) because knowledge of the clinical history is crucial for correct interpretation of the histologic findings in a skeletal muscle sample, most of which are not by themselves specifically diagnostic. Because muscle biopsy can help determine whether a patient has a neurogenic or a myogenic disorder, which sometimes is an issue that prompts performance of a biopsy, a brief discussion of clinical features and some laboratory findings in peripheral neuropathies is included.
The section about clinical features of neuromuscular disorders includes a brief discussion of the typical clinical presentation of myopathies and presents some information on the classic clinical presentations of a few different categories of myopathies, with some information about how these disorders are diagnosed.
It may be instructive to compare the structure and histology of normal skeletal muscle (see Skeletal Muscle - Structure and Histology) with the pathologic alterations observed in muscle (see Skeletal Muscle Pathology). The knowledge gained from this exercise can provide a basis for understanding the pathophysiology of some of these disorders, assist the reader to visualize the effect of disease at the tissue level, and provide insight into the process by which histopathologic diagnoses are formulated.
Indications
Key points:
-
Muscle biopsy is frequently necessary for diagnosis of myopathy.
-
Many types of myopathy cannot be reliably distinguished from each other without muscle biopsy.
-
Sometimes, muscle biopsy is even necessary in cases of myopathies caused by pathogenic mutations in known genes for which genetic testing is available.
-
If possible, treatment with immunosuppressive agents for suspected inflammatory myopathies should not be initiated until after muscle biopsy has been performed.
When a clinical diagnosis of myopathy is considered, muscle biopsy is often required for definitive diagnosis. For some exceptions to this requirement, see Myopathies That Might Not Require Muscle Biopsy, below.
Muscle biopsy is a fundamental component of the evaluation of a patient with possible muscle disease, also known as myopathy. At present, muscle biopsy is an essential part of the diagnostic investigation of patients whose disorders span most categories of muscle diseases, including inflammatory myopathies, toxic myopathies, many metabolic myopathies, some congenital myopathies, and many muscular dystrophies.
A major reason why muscle biopsy is often required for the diagnosis of neuromuscular disease is that there is considerable overlap in the clinical presentations of many different myopathies. Also, the clinical presentations of some myopathies and neuropathies can resemble each other, and there are similarities in the clinical presentations among many neurogenic processes. Therefore, different types of neuromuscular disorders cannot always be reliably distinguished from each other solely on the basis of the clinical features and results of laboratory studies.
A reason for performance of muscle biopsy that has emerged in this era of genetic testing is to determine the significance of genetic variants that are identified by such testing.
Concurrent with the proliferation of genetic tests and the widespread availability of genetic testing for neuromuscular diseases, ever-increasing numbers of genetic variants that are of uncertain clinical significance have been identified. These findings often necessitate performance of muscle biopsy after genetic testing has been performed to determine whether the genetic variants discovered by this testing are pathogenic and are responsible for an individual's neuromuscular disorder or are simply an unrelated finding or an epiphenomenon.
There are some hereditary neuromuscular disorders for which disease-modifying therapy is now available or is expected to become available, but still today, among myopathies, the inflammatory myopathies are the most common myopathies for which significantly effective therapies are available. [4, 5, 6]
Sometimes, a patient with an inflammatory myopathy is too seriously ill to allow a delay before initiating therapy; however, whenever possible, muscle biopsy should be performed before therapy is initiated, for the following reasons:
-
The risk of treatment, including steroids, other immunosuppressive agents, and, in some cases, intravenous immunoglobulin, which often must be administered for prolonged courses, is great enough that the diagnosis should be confirmed before therapy is started.
-
Many individuals who are thought to have inflammatory myopathy actually have other myopathic disorders for which immunosuppressive therapy is not indicated; therefore, it is best to avoid initiating potentially unnecessary therapy, if possible.
-
A delay often occurs between the initiation of therapy and a clinical response; to persevere with treatment in the absence of a prompt clinical response, it is beneficial to have complete confidence in the diagnosis before instituting therapy.
-
Treatment can alter the histopathologic findings; if treatment is started and the biopsy is subsequently performed because of a lack of clinical response to the therapy, it can be difficult to interpret the pathologic findings because the intervention may have altered them. Note: Treatment of only a few days or a week or two in duration will not usually significantly alter the biopsy findings in the inflammatory myopathies; nonetheless, whenever possible, it is best to avoid this potential complication by waiting to initiate therapy.
Repeat muscle biopsy is occasionally indicated for evaluation of the patient with known inflammatory myopathy who, after initial improvement with steroid or other immunosuppressive therapy, experiences increasing weakness. Biopsy findings can help distinguish between recurrence of the inflammatory disorder and steroid myopathy or other treatment-associated myopathic effect.
For disorders for which currently available therapeutic options are less definitive than those for inflammatory myopathies, a precise diagnosis is important for the following reasons:
-
Therapies that may be beneficial and may decrease symptoms or slow the progression of disease, though not curative, are available for some disorders.
-
Patients with certain disorders are eligible for therapeutic clinical trials.
-
Many myopathic disorders are hereditary diseases, and diagnosis is required for proper genetic counseling.
-
Patients often derive benefit and comfort from prognostic information; uncertainty about what is the cause of symptoms can be terrifying.
-
Biopsy can exclude the presence of another disorder for which there is specific therapy.
Occasionally, a muscle biopsy is indicated to distinguish between myopathy and neuropathy. The classic presentations of these two processes are clearly distinct; however, in practice, the clinical histories and physical and laboratory findings often overlap. Neuropathy and myopathy may also coexist, making a diagnosis based on clinical features alone particularly difficult or even impossible.
Myopathies That Might Not Require Muscle Biopsy
Key points:
-
Muscle biopsy may not be necessary for diagnosis of some myopathies that are caused by genetic alterations.
-
Because genetic testing is sometimes inconclusive, muscle biopsy may be necessary even in myopathies that are caused by genetic alterations.
-
Sometimes muscle biopsy is not necessary in individuals with endocrine myopathies.
-
If the clinical features and autoantibody profile are highly characteristic of a specific autoimmune disease, a muscle biopsy might not be necessary.
Several categories of myopathies do not typically require muscle biopsy for a diagnosis. This section discusses general circumstances in which a muscle biopsy is not always required for diagnosis of a myopathy and focuses on four categories of disorders for which muscle biopsy may be unnecessary.
The main exceptions to the requirement for muscle biopsy for accurate diagnosis of possible myopathy are for diagnosis of hereditary neuromuscular disorders that have characteristic clinical presentations and that are associated with known mutations in which genetic testing reveals genetic alterations that are known to be pathogenic and associated with the suspected disorders. In cases in which there is a family history of a well-characterized disorder and identified genetic alteration, if the individual undergoing evaluation has a similar clinical presentation, genetic testing will often suffice to make the diagnosis.
Because some of these hereditary myopathies do not exhibit specifically diagnostic features on muscle biopsy, muscle biopsy in these disorders will not yield a conclusive diagnosis and therefore may not be useful, though it may sometimes be needed to exclude the presence of another type of myopathy.
The following are examples of types of myopathies in which muscle biopsy may not be necessary:
-
Suspected dystrophinopathies, particularly when the clinical presentation has features that are highly characteristic of the dystrophies that are still commonly referred to as Duchenne or Becker muscular dystrophies
-
Myotonic dystrophies
-
Certain mitochondrial disorders
-
Certain metabolic disorders (the term metabolic disorders here refers to disorders of glycogen or lipid metabolism)
-
Periodic paralyses
In addition to many hereditary myopathies, muscle biopsy may not be necessary for the diagnosis of endocrine myopathies. Muscle biopsy may not always be necessary for diagnosis of some toxic myopathies and for certain other myopathic disorders not discussed here.
Category 1: genetic testing and muscular dystrophies, including dystrophinopathies
General issues, genetic testing in hereditary myopathies
Advances in molecular genetics have eliminated the need for muscle biopsy for many patients with dystrophinopathies and many other muscular dystrophies that have characteristic clinical presentations by permitting specific diagnosis of these disorders through genetic testing that is often performed on a sample of blood. Increasingly, however, the significance of some results of genetic testing is uncertain (see additional discussion of this below), necessitating the performance of a muscle biopsy after genetic testing has been performed.
Caution: Before you send a specimen for potentially costly genetic testing, please verify that the patient's insurance or another source will cover the cost or, if not, that the patient can bear the potentially substantial financial costs of these studies.
Multiple types of genetic tests are now available. These types include full sequencing (Sanger sequencing) of individual genes; Next Generation Sequencing (NGS) of groups of genes in parallel or for studying certain regions of genes; and Whole Exome Sequencing (WES), in which the entire protein-coding portion of the genome is sequenced. (In Whole Genome Sequencing, an individual's entire genome is sequenced, including both protein coding and noncoding regions; because of the vast amount of information obtained and challenges of interpretation, as well as the high cost, this latter process is still an emerging technology for clinical diagnosis, though it is sometimes performed.)
There are other genetic tests that can detect copy number variation and other genetic alterations. Detailed discussion is beyond the scope of this article.
There are a few main points about genetic testing for myopathies of which to be aware:
-
Different strategies for genetic testing can be used, depending on the specific clinical circumstances.
-
In situations where the clinical features suggest a single diagnosis, testing for a single gene (eg, dystrophin) can be performed.
-
If a small group of disorders is under consideration, with a limited group of genes that are likely candidates to be the cause of an individual's myopathy, targeted testing (NGS) can be performed for a select group of genes that can range from several to 100 or more in number.
-
WES can be performed as the initial genetic test or can be performed after targeted testing has not yielded a diagnosis.
-
The consequence of the availability of genetic testing is that in many cases, muscle biopsy will not be necessary.
-
Another consequence of this widespread and proliferating use of genetic testing is that results of uncertain significance are often obtained; muscle biopsy may then be necessary to determine whether the genetic alterations that have been uncovered are causally linked to the myopathies.
-
Another byproduct of this plethora of genetic testing is that there are many new findings demonstrating that the relationship between given mutations and the disorders they cause is complicated; for example, a single mutation in certain genes has been found to cause more than one type of myopathy, so that individuals with identical mutations sometimes can have different clinical disorders.
Genetic testing and dystrophinopathies
In patients with dystrophinopathies, mutations (most commonly deletions) can be demonstrated in the gene for dystrophin, located on the X-chromosome, locus Xp21. This extremely large gene, comprising 2 million base pairs, codes for a structural protein of skeletal muscle located on the internal surface of the sarcolemma. Sarcolemma is the term for the plasma membrane of a muscle cell, also known as a myofiber.
Until relatively recently, the large size of the dystrophin gene precluded searching the entire gene for point mutations; however, dystrophin gene sequencing is now commercially available and is routinely performed. Some laboratories first test for the common deletions and duplications in the dystrophin gene that are known to be pathogenic and the cause of some dystrophinopathies and then sequence the entire gene if the initial deletion/duplication studies are negative.
Dystrophinopathies most commonly present in young boys in the preschool and elementary-school age range. Among individuals ultimately diagnosed with a dystrophinopathy, muscle biopsy now is usually performed only in patients with clinical syndromes that differ from typical dystrophinopathies (eg, adults with progressive limb-girdle syndromes or certain other unique or unusual clinical circumstances).
This category includes some females who have one X-chromosome with a wild-type (normal) dystrophin gene but whose other X-chromosome contains a dystrophin mutation, in whom an unfortunate pattern of random inactivation of X-chromosomes gives rise to clinical myopathy because the number of myofibers (muscle cells) expressing the mutant dystrophin gene is great enough to produce symptomatic myopathy.
In some of the cases of individuals suspected to have a dystrophinopathy, there will be a family history of a dystrophinopathy, and in this situation, even if an individual does not exhibit one of the classical syndromes of dystrophinopathies, muscle biopsy may be avoided. Some cases of dystrophinopathies are due to de-novomutations in individuals, and thus there will be no family history in these cases that could serve to raise the index of suspicion for these disorders.
Genetic testing instead of muscle biopsy in some other myopathies
Genetic testing of blood samples is available for abnormalities of genes for other muscle membrane, structural, and myofibrillar-associated proteins that can present as limb-girdle syndromes, such as any of the four sarcoglycans, dysferlin, caveolin-3, calpain, lamin A/C, fukutin-related protein, and, now, essentially any genetically defined myopathy. [9, 8]
Sometimes, muscle biopsy is still performed first to narrow the diagnostic possibilities, often by excluding an inflammatory myopathy, and is then followed by directed genetic testing if an alternate specific diagnosis is not identified on muscle biopsy. With increasing frequency, genetic testing is performed first, and if this is unrevealing or if the significance of the results obtained is uncertain, muscle biopsy is then performed.
If a suspected myopathy does not exhibit specific pathologic findings on muscle biopsy, genetic testing may be performed, rather than muscle biopsy, which is unlikely to yield a specific diagnosis. For example, genetic testing is available for facioscapulohumeral dystrophy and Perlecan deficiency (Schwartz-Jampel syndrome). Muscle biopsy in these disorders demonstrates only nonspecific myopathic findings and therefore is generally not indicated for diagnosis of these disorders. There are numerous other hereditary myopathies with characteristic clinical presentations for which genetic testing can be performed that can obviate the need for muscle biopsy in these cases.
When genetic testing in an individual suspected of having a hereditary myopathy reveals a mutation that is known to be pathogenic and that fits with the individual's clinical syndrome, muscle biopsy is not needed. Unfortunately, sometimes the results of genetic testing are not definitive, and muscle biopsy might then be indicated in cases with negative or ambiguous results from genetic testing.
It's complicated
The current widespread use of panels of genetic tests for a large number of muscular dystrophy and other myopathy-associated genes [10] has led to a new problem, which is the uncovering of variants of unknown significance (VOUS or VUS) in a gene or genes. Muscle biopsy may be needed to determine if a given VUS is myopathic. For example, if genetic testing of an individual reveals a VUS in the gene for dystrophin, muscle biopsy can be performed to look for myopathic features in the muscle and to establish whether there is normal or abnormal expression of the dystrophin protein in the muscle, thereby determining whether the VUS might be the underlying cause of a myopathy.
Sometimes, genetic testing reveals more than one genetic alteration, and it is not known which one is responsible for the individual's myopathy. In this circumstance, muscle biopsy might provide the specific diagnosis.
Thus, despite the great strides that are being made in elucidating the molecular biology of many myopathies, the performance of genetic testing does not always eliminate the need for muscle biopsy.
Category 2: myotonic dystrophies
Myotonic dystrophy type 1 is definitively diagnosed by means of genetic testing on a sample of blood, which reveals a characteristic increase in the number of CTG triplet repeats in the gene for muscle protein kinase on chromosome 19. Myotonic dystrophy type 2 (proximal myotonic myopathy or PROMM) is due to increased CCTG quadruplet repeats in a zinc finger protein gene on chromosome 3, also detectable by testing on a sample of blood.
Athough some of the histopathologic findings on muscle biopsy in these disorders are fairly characteristic, they are not specifically diagnostic; thus, if these disorders are suspected on the basis of the clinical presentation, genetic testing of blood, rather than muscle biopsy, is indicated.
Category 3: periodic paralyses
Periodic paralyses are uncommon disorders that are caused by mutations in various genes for muscle membrane ion channels. These disorders have unique clinical, biochemical, and electrodiagnostic features. Because they lack specifically diagnostic findings on muscle biopsy, genetic testing is the preferred way to confirm the suspected diagnosis of one of these disorders.
Dilatation of the T-tubule system is found in some patients with hypokalemic periodic paralysis, which produces vacuoles in histologic sections, but this finding is not specific enough to be diagnostic. Muscle biopsy can also demonstrate a nonspecific myopathic picture in these disorders. See Skeletal Muscle Pathology for examples of nonspecific myopathic features.
Category 4: endocrine myopathies
Myopathy can be a feature of disorders of thyroid, parathyroid, and adrenal function. The favored way to diagnose endocrine myopathies is to recognize their clinical presentations and perform serologic testing for appropriate components of the hypothalamic-pituitary-endocrine organ axis. The myopathic symptoms in an individual with endocrine myopathy should resolve after the endocrine disorder is treated. (If significant myofiber loss and/or fibrosis has already developed, the myopathic symptoms might not fully resolve upon treatment of the endocrine disorder.)
Myotonic dystrophy, periodic paralyses, and endocrine myopathies are not considered further in this article. More information about myotonic dystrophy is available from the Myotonic Dystrophy Organization.
Anesthesia
Open muscle biopsy is performed with local anesthesia for most adult patients. General anesthesia is typically required for infants and young children.
If surgery is performed under local anesthesia, care should be taken not to inject the anesthetic directly into the biopsy site. This minimizes the risk of introducing a track from the needle and the anesthetic into the muscle, which can interfere with interpretation of the biopsy. The local anesthetic should be injected just under the skin in an oval area, whose length is approximately 1.5 times that of the incision.
Percutaneous needle biopsies are not discussed here.
Clinical Features of Neuromuscular Disorders
Key points:
-
Most individual histopathologic features in muscle biopsies are not by themselves diagnostically specific, so knowledge of the details of the clinical features is often needed to determine the significance of the biopsy findings.
-
The most common clinical hallmark of any category of myopathy is weakness of proximal muscle groups, so it can be difficult to distinguish different myopathies based only upon clinical features.
-
Atypical clinical presentations of some forms of myopathy and neuropathy can make it difficult to reliably determine whether an individual has a myopathic or neuropathic disorder.
-
Some metabolic myopathies are characterized by fluctuation of muscle power during activity.
-
Individual inflammatory myopathies exhibit characteristic features, including age of onset, tempo of progression, specific distribution of affected muscle groups, involvement of other organ systems, and specific autoantibody profile.
-
The creatine kinase level is an important blood test to obtain for evaluation of suspected myopathy.
Importance of provision of clinical information for interpretation of a muscle biopsy
Few findings in a muscle biopsy are pathognomonic for a specific diagnosis. Instead, a typical muscle biopsy sample presents a constellation of findings that by themselves could have multiple potential interpretations and that, ideally, should be analyzed within the context of the clinical features of the individual case. In other words, to be able to properly assess the significance of histologic findings in a particular muscle biopsy sample, the pathologist must have information about the clinical presentation of a given patient.
As an example of the necessity of clinical information to determine the significance of histopathologic findings, consider the finding of clear cytoplasmic vacuoles in hematoxylin and eosin (H&E) cryostat sections of muscle biopsies. The most common reason for their presence is technical artifact due to ice crystal formation when a tissue sample is frozen for the preparation of cryostat sections, in which case the vacuoles are diagnostically insignificant. However, they are also present in many myopathic disorders, including but not limited to certain glycogen storage diseases, lipid myopathies, periodic paralyses, and toxic myopathies that can result from treatment with colchicine, chloroquine, or amiodarone.
Knowledge of the clinical history makes it possible for the pathologist to decide which diagnostic considerations are reasonable in a given case and assists in the determination of whether additional special studies are indicated. For example, in the case of clear vacuoles, if a biopsy shows only a few myofibers with vacuoles, the pathologist must decide whether the vacuoles are insignificant technical effects or whether they are the key diagnostic finding. The details of the clinical history provide guidance for the pathologist in interpreting the significance of the finding.
Interpretation of a muscle biopsy is similar to the process of clinical diagnosis, in which one synthesizes all of the information available about an individual patient to arrive at the correct diagnosis. If you are ever involved in requesting or performing a muscle biopsy for a patient, please allow the muscle pathologist to fully perform his/her/their function by providing reasonably detailed clinical information that can permit this physician to fully interpret biopsy findings that are not, by themselves, diagnostically specific. Provision of this information is not for the benefit of the pathologist but for the benefit of the patient, the human being undergoing a surgical procedure with the hope of finding the cause of his/her/their symptoms.
Against this background, the technical issues that must be addressed by the physicians involved with muscle biopsies are as follows:
-
Proper selection of a muscle for biopsy
-
Biopsy procedure and immediate handling of the tissue in the operating room
-
Studies performed on the biopsy sample
These considerations are addressed in more detail below (see Technical Issues in Muscle Biopsy).
Signs, symptoms, and initial laboratory testing of suspected neuromuscular disorders
Signs and symptoms
Disease of skeletal muscle tissue can be expressed in very few ways, as follows:
-
Weakness or decreased movement
-
Muscle ache, also referred to as myalgia
-
Cramps
-
Abnormal changes in strength as a result of physical activity
The main clinical hallmark of neuromuscular disease, whether of neurogenic or of myopathic origin, is weakness. Weakness is manifested in age-related variations. For example, weakness in utero can be expressed as decreased fetal movements and may be recognized by a woman who has had previous pregnancies.
In the neonatal period, the infant who has a myopathy may be hypotonic; this is termed the floppy infant syndrome. Neuromuscular disease is only one of a number of potential causes of this syndrome. In later infancy and during the toddler years, delay in acquisition of motor-developmental milestones is typically a major sign of myopathy. From childhood through adulthood, diminished muscle power, reported by affected patients and ascertained by direct testing of the strength of individual muscle groups, is in many cases a characteristic clinical feature of neuromuscular disease.
The classic clinical signs of myopathy include the following:
-
Weakness, which usually affects the proximal muscle groups, most often the shoulder and limb girdles; weakness of axial muscles can also be identified in some disorders.
-
Preservation of muscle-stretch reflexes until relatively late in the course of the disorder.
-
Absence of abnormalities of somatosensation.
Variation of strength with activity can occur in some patients with muscle disease. This can mean either decremental or, rarely, incremental change in strength with a degree of activity that would not result in this change in a healthy individual.
McArdle disease, a metabolic myopathy
Fluctuation of muscle power or symptoms that develop during exercise should raise suspicion for the presence of a metabolic myopathy. For example, in McArdle disease, myophosphorylase deficiency leads to inability to mobilize the glycogen needed for muscular activity for movement or for development of isometric tension needed to hold or carry heavy objects.
A patient with McArdle disease has pain, sometimes cramps, and weakness during the anaerobic phase of exercise. If the patient can exercise at a low level during the anaerobic phase to avoid drawing on glycogen stores, once the aerobic phase is finally reached and glycogenolysis is no longer needed because free fatty acids can be used for generation of energy, the patient's performance improves and symptoms can be avoided. This improvement is referred to as the second wind phenomenon.
In addition to a switch to reliance on free fatty acids when glycogen cannot be adequately mobilized, during the aerobic phase of energy metabolism, more energy is produced per unit of glucose metabolized, and therefore less glycogen is needed and less glucose is required to generate more energy than is produced by anaerobic metabolism.
Myasthenia gravis
Fatigability denotes progressive loss of muscle power with exertion that improves with rest. This is often a defining clinical feature of myasthenia gravis, a disorder of impaired neuromuscular transmission. Muscle biopsy is typically not performed if the presentation is characteristic of myasthenia gravis. Myasthenia gravis is usually diagnosed on the basis of the following:
-
Clinical presentation, typically characterized by abnormal fatigability, which can be worse later in the day
-
Electrodiagnostic studies showing a decremental response in the muscle with repetitive stimulation of the motor nerve
-
Presence of serum antibodies to the acetylcholine receptor or to certain other serum antibodies known to be associated with this disorder
Inflammatory myopathies
Idiopathic inflammatory myopathies, [5, 4] which are autoimmune diseases, typically have a subacute presentation, in which symptoms develop over a period of weeks to a few months. In most of the idiopathic inflammatory myopathies, proximal muscle groups are affected and distal muscle groups are relatively spared. One type, sporadic inclusion body myositis, often also affects distal muscles, particularly finger flexors, and the presentation in this disorder is often more indolent than in other inflammatory myopathies, progressing insidiously over a longer period of time, sometimes over multiple years.
The idiopathic inflammatory myopathies as a group occur across a wide spectrum of age groups; there is some age-related variation of subcategories. Dermatomyositis, which can occur in both adults and children, is the most common inflammatory myopathy that occurs in children, when it can be referred to as juvenile dermatomyositis. Inclusion body myositis tends to occur in people older than 50 years and affects more men than other types of inflammatory myopathies, which tend to have a female predominance.
In some subcategories of inflammatory myopathies, there will be evidence of autoimmune disease of other organ systems, and in addition to the inflammatory myopathies, there may be characteristic dermatologic disorders, arthritis, or cardiac, pulmonary, renal, or gastrointestinal disease. Autoantibodies are present in many individuals with inflammatory myopathies. Different subtypes of inflammatory myopathies, and autoimmune disorders in general, are associated with different autoantibodies. [4, 5, 11]
Myalgia is present in some patients with inflammatory myopathy. This symptom sometimes is present in noninflammatory disorders in which there is necrosis of myofibers. Myalgia is also found in some patients with metabolic diseases that affect skeletal muscle and occurs when the energy supply of the muscle is depleted and lactic acid builds up; cramps sometimes occur.
Neurogenic disorders
In contrast to myopathy, the classic clinical features of peripheral neuropathy include the following:
-
Weakness, predominantly affecting distal musculature
-
Decrease of muscle-stretch reflexes, particularly in demyelinating neuropathies
-
Fasciculations, when abnormal excitability of the motor neuron is present
-
Concomitant somatosensory abnormalities, which occur in many peripheral neuropathies, though these are not present in pure motor neuropathies
In their conventional clinical presentations, distinguishing muscle disease from peripheral nerve disease is a straightforward matter. In practice, however, this distinction is not always simple. There are several reasons why it may be difficult to determine whether a patient has neuropathy or myopathy on clinical evaluation:
-
One reason is that some myopathies affect distal muscles; myotonic dystrophy, sporadic inclusion body myositis (sIBM), hereditary IBM, some of the myofibrillar myopathies, and several other distal myopathies can all affect distal muscle groups.
-
Some neurogenic disorders, including diabetic amyotrophy and motor neuron disease, may affect proximal muscles.
-
Some patients may have combined neurogenic and myopathic disorders. For example, a patient with diabetic neuropathy can also acquire an inflammatory myopathy, or a patient who has peripheral neuropathy caused by chemotherapy for cancer may develop dermatomyositis as a paraneoplastic syndrome or other paraneoplastic inflammatory myopathy, or a patient can have both radiculopathy caused by degenerative joint disease in the vertebral column and a primary myopathy. In these examples, the clinical findings are complicated, which can make it difficult to identify the correct diagnostic category solely on the basis of the clinical features.
For an excellent and more detailed discussion of clinical evaluation of patients with neuromuscular disease that is beyond the scope of this article, see chapter 1, "Approach to patients with neuromuscular disease," in the second edition of Neuromuscular Disorders, by Anthony Amato and James Russell. [12]
A very old but nonetheless superb and well-written monograph by Michael H Brooke, A Clinician's View of Neuromuscular Diseases, provides insight into the clinical assessment of patients with neuromuscular disease. [13] Although it was written before the explosion of information regarding the molecular genetics of neuromuscular disease, it remains a unique and valuable tool. It is out of print, but copies can still be found for purchase and in libraries.
Laboratory studies
The serum creatine kinase (CK) level is the single most important blood value to obtain when myopathy is being considered. A representative normal reference range from one laboratory is 24-196 international units per liter (IU/L); each laboratory has its own reference range, with the upper limit of normal usually below 250 IU/L. The CK level is useful, but not definitive, in determining whether myopathy is present and in distinguishing between neuropathy or myopathy.
Moderately to extremely elevated levels of CK (>1500 IU/L) usually indicate muscle disease. Mildly elevated levels (200-800 IU/L) can be observed in either neurogenic or myopathic disorders. Normal levels are not often found in the patient with myopathy, but patients with myopathy who have reduced residual muscle mass may have serum CK levels that fall within the normal range. Conversely, patients with substantial muscle mass may have CK levels above the normal range in the absence of disease; therefore, a CK level of, for example, 500 IU/L does not always indicate the presence of myopathy.
The serum aldolase level can be helpful in providing evidence of myopathy. An abnormal aldolase level is less specific for skeletal muscle disease than the CK level is, but because of its longer half-life in serum, the serum aldolase level is sometimes elevated in the setting of myopathy when the CK level is normal. If the CK level is already known to be abnormal, there is no need to obtain testing for the aldolase level.
Electrodiagnostic studies
Electrodiagnostic studies are often extremely useful in determining whether a neuropathic, myopathic, or mixed disorder is present.
Changes in nerve conduction velocities, the compound muscle-action potential, or both can be present in neurogenic disorders.
Electromyography (EMG) demonstrates different findings in neurogenic and myopathic disorders and can be useful to help distinguish them; specific details are beyond the scope of this article. Avoiding EMG in a muscle that will undergo biopsy is of critical importance. EMG inflicts damage on the muscle that can interfere with proper interpretation of a biopsy for 1-2 months. For patients with suspected myopathy, needle EMG should be performed on muscles on only one side of the body; a subsequent muscle biopsy should be performed on the other side.
Technical Issues in Muscle Biopsy
Selection of muscle for biopsy
Biopsy of a clinically involved muscle is important. Most disorders preferentially or more severely affect certain muscles while relatively or completely sparing other muscles.
To increase the likelihood of sampling the pathologic process, it is important to select a symptomatic muscle. Selection of a muscle should also be based on the expected distribution of the leading clinical diagnosis. For example, if the leading diagnostic consideration is polymyositis, a proximal muscle, such as the vastus lateralis of the quadriceps, is usually the best choice for biopsy.
-
Please be aware that some disease processes have a patchy distribution rather than being diffusely distributed throughout the affected muscles; thus, even if an affected muscle is biopsied, occasionally the biopsy might not sample the pathology.
When possible, biopsy should be performed on a mildly symptomatic muscle, equivalent to power of 4/5 on the Medical Research Council scale used to grade strength. Biopsy of a muscle that is too weak or atrophic may result in obtaining a sample of end-stage muscle (see the first image below). In end-stage muscle, loss of myofibers is severe, and the myofibers are replaced by fibrovascular and adipose tissue, typically without residual clues to the process that caused the muscle damage. On occasion, only the presence of a residual muscle spindle confirms that the specimen is a biopsy sample of skeletal muscle (see the second image below).
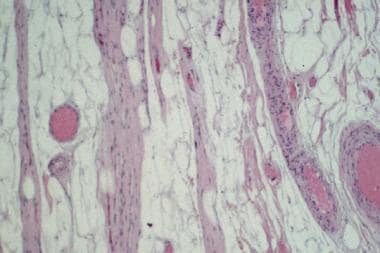 Hematoxylin-eosin (H&E) paraffin section of a muscle biopsy sample reveals end-stage muscle. Fibrovascular and adipose tissues have entirely replaced this muscle, which can therefore provide no information about patient's underlying pathologic process.
Hematoxylin-eosin (H&E) paraffin section of a muscle biopsy sample reveals end-stage muscle. Fibrovascular and adipose tissues have entirely replaced this muscle, which can therefore provide no information about patient's underlying pathologic process.
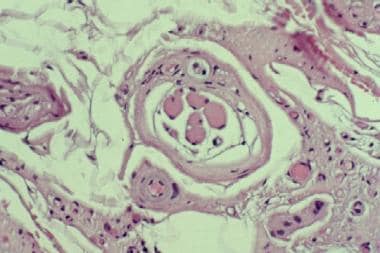 Hematoxylin and eosin (H&E) paraffin section of this muscle biopsy sample reveals end-stage muscle. The structure in center of this image, consisting of cluster of small muscle fibers surrounded by a fibrous capsule, is a muscle spindle; this is the only finding that confirms the identity of this specimen as skeletal muscle.
Hematoxylin and eosin (H&E) paraffin section of this muscle biopsy sample reveals end-stage muscle. The structure in center of this image, consisting of cluster of small muscle fibers surrounded by a fibrous capsule, is a muscle spindle; this is the only finding that confirms the identity of this specimen as skeletal muscle.
Biopsy procedure and immediate tissue handling
In performing a muscle biopsy, it is essential to cause as little trauma to the muscle tissue as possible so as to reduce the risk of disrupting the muscle architecture and minimize the risk of introducing contraction band artifact and other technical artifacts. Electrocautery should not be used to obtain a specimen for muscle biopsy.
The two images below demonstrate effects of the application of electrocautery to skeletal muscle during biopsy. The first shows an area of tissue coagulation that results from application of electrocautery, and the second shows severe contraction band artifact caused by the electric current; both of these technical effects interfere with interpretation of a muscle biopsy, particularly if they are widespread.
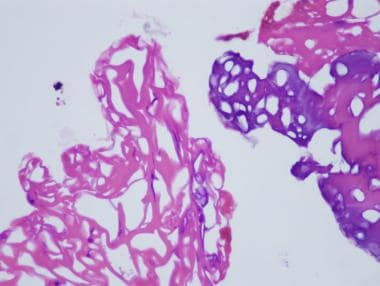 Hematoxylin and eosin paraffin section of a region of a skeletal muscle sample that was coagulated by application of electrocautery to tissue during biopsy procedure. This tissue is unrecognizable.
Hematoxylin and eosin paraffin section of a region of a skeletal muscle sample that was coagulated by application of electrocautery to tissue during biopsy procedure. This tissue is unrecognizable.
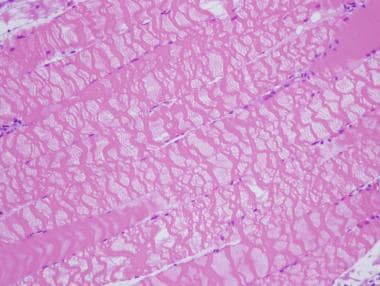 This hematoxylin and eosin (H&E) paraffin section shows severe contraction band artifact in the myofibers in this area of muscle biopsy, which resulted from application of electrocautery. This area of sample is not coagulated, as seen in previous image from a different region of this biopsy, see above image, and thus can be recognized as skeletal muscle, but there is severe disruption of architecture of affected myofibers, which can significantly interfere with interpretation of muscle biopsy.
This hematoxylin and eosin (H&E) paraffin section shows severe contraction band artifact in the myofibers in this area of muscle biopsy, which resulted from application of electrocautery. This area of sample is not coagulated, as seen in previous image from a different region of this biopsy, see above image, and thus can be recognized as skeletal muscle, but there is severe disruption of architecture of affected myofibers, which can significantly interfere with interpretation of muscle biopsy.
Electrocautery can also cause lack of staining of myofibers (myofibers are muscle cells) with the stains that are performed on cryostat (frozen) sections of muscle, particularly the enzyme histochemical stains that are frequently performed on muscle biopsies. Examples of some of these stains are found in Skeletal Muscle - Structure and Histology and Skeletal Muscle Pathology.
The specimens required and the details of the preferred method of biopsy handling vary among medical centers and commercial laboratories that process muscle biopsies. It is essential to consult the laboratory at the center that will receive the biopsy sample so as to learn the requirements and the preferred method of handling and, when relevant, shipping the tissue. Ultimately, of course, the surgeon must determine the precise surgical method for each patient, but he/she/they should perform the procedure with knowledge of what is required by the laboratory where the specimen will be processed.
The information below is an example of the procedure adapted for one specific medical center and should be considered only a general guide. These considerations should be tailored to meet the needs of the individual patient and institution.
Please note that these procedures cannot be used for evaluation of a biopsy for malignant hyperthermia; if this disorder is under consideration, please consult the website of the Malignant Hyperthermia Association of the United States, https://www.mhaus.org.
The typical muscle biopsy sample consists of two specimens: fresh and fixed. In some laboratories, a specimen for electron microscopy is taken from the fixed specimen; other laboratories request a separate additional sample of muscle be submitted for electron microscopy.
In certain special clinical circumstances, an additional special sample for biochemical analysis or special genetic testing may contribute to the specific diagnosis. Today, the most common use of this special sample of muscle is for assay of the activities of mitochondrial enzymes; this assay is performed by only a limited number of laboratories and is indicated in only select clinical circumstances. This sample is also suitable for genetic testing performed by certain laboratories.
On occasion, a muscle biopsy sample consists only of a single fresh specimen, usually in the form of multiple minute fragments, obtained by means of needle biopsy. In one center, with a strong research program in muscle disease, a strong motivation to perfect the technique of needle biopsy, and physicians experienced with the procedure, needle biopsy is the preferred method of muscle biopsy, with a diagnostic yield reportedly comparable to that of open biopsy [14] ; however, a full open biopsy is preferred by most medical centers and laboratories that process muscle biopsies.
Some laboratories simply do not have the level of technical expertise required to adequately process and section tiny needle biopsies. In these very small limited specimens, it is difficult to find areas where it is possible to observe patterns of pathology and distribution of pathology, which can yield diagnostic information.
Needle biopsy generally provides a specimen of limited size that typically is not well oriented; however, it may be the method of choice under the following circumstances:
-
When serial biopsy procedures are required to follow the course of the disease or to monitor the response to therapy in a patient
-
When a disease with diffuse distribution is the leading diagnostic consideration, so that any sample of tissue is likely to be pathologic
-
When a small sample of muscle is needed only for biochemical assay or a certain genetic test
-
When a patient will not consent to an open biopsy but will allow a needle biopsy
-
When open biopsy is contraindicated
A fresh muscle biopsy specimen (see the image below) is used for histochemical studies and certain immunohistochemistry studies that are optimized for cryostat sections of tissue and is sometimes used for immunofluorescence in selected cases, when indicated, by laboratories that are set up to perform immunofluorescence. The fresh specimen should measure approximately 0.5 × 0.5 cm in cross-section, or 0.5 cm in diameter, and 1 cm in length along the longitudinal axis of the muscle fibers.
 A fresh muscle biopsy specimen is mounted on cork, in this case using gum tragacanth as the mounting medium. The tissue sample is poised above vial of isopentane, which is chilled with liquid nitrogen coolant. The specimen will be frozen by gently immersing it in the isopentane. In this photograph, myofibers are oriented longitudinally in vertical plane. The tissue will be sectioned by using a cryostat microtome to produce cross-sections of skeletal muscle tissue.
A fresh muscle biopsy specimen is mounted on cork, in this case using gum tragacanth as the mounting medium. The tissue sample is poised above vial of isopentane, which is chilled with liquid nitrogen coolant. The specimen will be frozen by gently immersing it in the isopentane. In this photograph, myofibers are oriented longitudinally in vertical plane. The tissue will be sectioned by using a cryostat microtome to produce cross-sections of skeletal muscle tissue.
The fresh sample can be sent to the laboratory on saline-moistened gauze in a sealed container on ice (regular ice, not dry ice); in this way, the tissue stays chilled without becoming frozen. The tissue should not be immersed (ie, should not be floating or drenched) in sodium chloride solution, because this will lead to the formation of ice crystals in the myofibers when the sample is frozen, which will appear as vacuoles. Some laboratories use specific transport media for the fresh specimens.
When the specimen arrives in the laboratory, the technologist mounts it using an appropriate mounting medium, with the myofibers in the appropriate orientation and snap-freezes the specimen in isopentane that is chilled in liquid nitrogen. Frozen sections are cut from this sample on a cryostat, which is a microtome that is maintained at temperatures below freezing. (The terms frozen sections and cryostat sections are interchangeable.)
Optimally, after the biopsy procedure, this fresh specimen is immediately transported to the laboratory for processing to prevent the tissue from losing any of its enzymatic reactivity, which is needed for histochemical studies, or immunogenicity, which is needed for immunohistochemical studies. In most situations, however, the sample remains in satisfactory condition for most necessary studies if refrigerated overnight or even if refrigerated for a few days in the event of an unavoidable delay, although a delay of longer than overnight is definitely not recommended. If it is not refrigerated, the tissue will lose enzymatic reactivity and will, ultimately, undergo decompositional change.
A fixed specimen (see the image below) is used for routine microscopy and possible electron microscopy (EM). As mentioned above, some laboratories request that a separate additional sample be obtained for EM and placed in a fixative that is appropriate for EM at the time of biopsy.
-
EM is reserved for special situations in which it is expected to substantially contribute to the diagnosis, on the basis of the clinical history, the light-microscopic histopathologic findings in the cryostat and paraffin sections, or both.
The fixed specimen should have dimensions similar to those of the fresh specimen. It must be handled properly to maintain orientation of the myofibers, to keep the myofibers at rest length, and to prevent contraction of the myofibers.
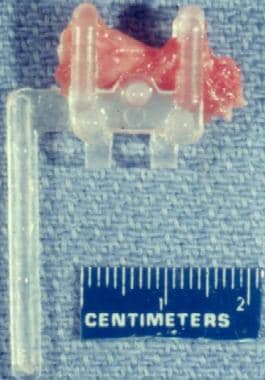 Specimen of skeletal muscle on a 10-mm Rayport clamp is fixed in paraformaldehyde. The longitudinal axis of fibers is oriented in horizontal plane. (A small stray piece of muscle that is not clamped lies obliquely over the clamped portion of specimen.)
Specimen of skeletal muscle on a 10-mm Rayport clamp is fixed in paraformaldehyde. The longitudinal axis of fibers is oriented in horizontal plane. (A small stray piece of muscle that is not clamped lies obliquely over the clamped portion of specimen.)
The sample is optimally removed from the patient by using a special clamp designed for this purpose, such as the 10-mm Rayport clamp (V. Mueller, McGaw Park, IL, available from multiple distributors; see the image above). There are other muscle clamps that may be preferred by some surgeons or laboratories. This biopsy sample is obtained in the following manner:
-
A segment of muscle of the desired dimensions is dissected; the bottom portion of the clamp is inserted below this segment of muscle in the posts-up position so that the length of the fibers runs perpendicular to the jaws of the clamp.
-
After the bottom portion of the clamp is inserted, the top portion of the Rayport clamp can be folded over and the holes fitted onto the posts; the surgeon then excises the muscle fibers 1-2 mm external to the clamp, and the specimen is placed in fixative (the preferred fixative in some laboratories is 4% paraformaldehyde).
If a special clamp is not available for the procedure, alternative methods of obtaining the fixed specimen are available. The sample can be sent as a fresh specimen to the laboratory, where the technologists perform the procedures needed for immobilization and fixation. Another method involves suturing or pinning the specimen to a tongue blade or a piece of cork for immobilization prior to fixation.
If paraformaldehyde is not available, 10% neutral buffered formalin is an acceptable alternative for the purpose of light microscopy. If, however, EM is needed, the specimen initially fixed in paraformaldehyde has better ultrastructural preservation than that of a sample fixed in formalin, though a few hours of formalin fixation prior to placement of the sample in a fixative appropriate for EM should not interfere with this study.
If paraformaldehyde is not available and it is anticipated that EM will be needed, a small portion of muscle can be placed directly in 3% glutaraldehyde (or other EM-appropriate fixative, such as Karnovsky's fixative) at the time of biopsy for submission to the EM laboratory. To prevent contraction of the muscle, the sample should be immobilized at rest length before it is immersed in the fixative. The specimen to be immersed must be small (1-2 mm wide or deep) because glutaraldehyde penetrates tissue slowly. If the specimen is too large, portions of the sample will not be adequately fixed for EM before they begin to become degraded.
After overnight fixation in paraformaldehyde, the technologist separates a small section and submits it in glutaraldehyde for embedment for EM. The remainder is submitted for paraffin processing, with the end(s) of the specimen removed and placed in cross-section and the rest of the sample submitted in longitudinal section.
An additional fresh specimen may be useful in selected cases where the presence of a metabolic myopathy, particularly a mitochondrial disorder, is strongly suspected. The sample may be sent to specialized laboratories for assessment of specific enzymatic activities, such as for assay of the activities of mitochondrial enzymes. Such a sample can be used for measurement and characterization of specific protein constituents in muscle (eg, the protein dystrophin), but in most cases, the availability of genetic testing on a sample of blood has made Western blot analysis of muscle proteins unnecessary, and this type of study is not frequently done today.
This additional special specimen should be of dimensions similar to those of the other specimens, and optimally, it should be snap-frozen in liquid nitrogen at the location of the procedure, or promptly be brought to the laboratory for immediate freezing, because of the lability of some of these cellular constituents. It should be stored in a freezer at –70°C. Laboratory personnel should be alerted in advance if the need for this type of specimen is anticipated. Many medical centers are not equipped to perform this service.
If liquid nitrogen is not available, this extra sample can also be immediately frozen by using dry ice. Care should be taken to insulate this sample from the rest of the biopsy when the specimen is being transported to the laboratory, so that the other parts of the biopsy do not become frozen.
Studies performed on the biopsy sample
For examples of these studies in normal skeletal muscle, see Skeletal Muscle - Structure and Histology; for examples of these studies in various neuromuscular disorders, see Skeletal Muscle Pathology.
The actual methods for performing the stains can be found in standard histology texts (books or online) and pathology laboratory manuals. Immunohistochemical stains must be performed by a laboratory set up for this purpose. The manufacturer provides instructions for use of each individual antibody, and each laboratory develops its own modifications to achieve optimal results with the stains.
There are multiple histologic, histochemical, and immunohistochemical stains that are performed on muscle biopsies. Each institution that processes muscle biopsies has a select battery of stains from among these stains that is routinely performed on the frozen sample in addition to the H&E stain. This routine panel of stains assists in screening for and assessing neurogenic or other types of atrophy, inflammatory myopathies, noninflammatory myopathic disorders, congenital myopathies, and metabolic diseases, as well as in demonstrating structural changes or inclusions that are diagnostic of certain specific disorders.
Some of these studies cannot be performed on material that has been fixed and embedded in paraffin. Some of these stains, because they utilize the activity of enzymes within the myofibers, require fresh/frozen tissue and therefore cannot be performed on fixed tissue. Some of the structures and materials are dissolved by paraffin processing and thus can only be identified in frozen sections.
After review of the initial battery of stains, if the composite of clinical and pathologic findings warrants it, the pathologist may decide to perform additional special studies.
An example of one battery of stains performed on every biopsy by one institution includes those listed below. The routine panel of stains varies between different laboratories.
H&E is the routine histologic stain used for evaluation of basic tissue organization and cellular structure.
With nicotinamide adenine dinucleotide tetrazolium reductase (NADH) staining, the activity of this group of enzymes is demonstrated by the transfer of hydrogen to a compound that turns gray-blue when it is reduced. These enzymes are found in mitochondria and endoplasmic reticulum. This stain is used to assist in evaluating for neurogenic atrophy, mitochondrial disorders, and central core disease, among others, and it is useful for detecting subtle alterations of intracellular structure in a myofiber that suggest it is not well.
Enzyme histochemistry for the combined activities of two mitochondrial enzymes, succinic dehydrogenase and cytochrome oxidase, is performed to assess for mitochondrial function. The succinic dehydrogenase stain is blue, and the cytochrome oxidase stain is golden brown. When the activity of both of these enzymes is present, the myofibers will be the combination of blue plus gold—in other words, taupe in color. Myofibers that lack cytochrome oxidase activity will lack the gold staining from the activity of this enzyme and will thus appear blue.
Fiber-typing stains are also used. Muscle is composed of two main myofiber types: 1 and 2. Some disease processes characteristically affect one type or the other, resulting in atrophy of either type 1 or type 2 myofibers. Other processes, such as neurogenic disorders, can alter the distribution of myofiber types. Some laboratories use a myosin adenosine triphosphatase (ATPase) hitochemical stain at multiple pH levels to demonstrate the different fiber types. This is a labor-intensive stain to perform.
Immunohistochemistry is an alternative method for identifying and demonstrating myofiber types in muscle biopsies. This automated procedure can be performed for the different forms of myosin heavy chains found in type 1 and type 2 myofibers. These stains are adequate for myofiber typing in most cases. (Novocastra [Newcastle upon Tyne, England] recommends an immunohistochemical stain for research purposes only.) Stains are now also available for embryonic myosin heavy chain as well as other subtypes of myosin heavy chains. Immunohistochemical stains are now available for different forms of myosin ATPase.
The modified Gomori trichrome stain is particularly helpful in evaluating for the presence of mitochondrial disorders, inclusion body myositis, and nemaline myopathy, among many other uses.
Periodic acid–Schiff (PAS) stains glycogen and other polysaccharides. This stain is most useful for the diagnosis of glycogen storage diseases. PAS also stains the basal lamina of blood vessel walls, so it can be useful for evaluating the structure of vessels.
Fat stains, including Sudan Black, oil-red-O, and osmium, are used to demonstrate the presence of neutral lipids in muscle, which are normally present but can exist in abnormal amounts or distribution in carnitine deficiency, some mitochondrial disorders, acquired metabolic disorders (eg, in starvation), and nonspecific abnormalities of the myofibers.
Human leukocyte antigen (HLA) class ABC by immunohistochemistry is used to identify and support the diagnosis of autoimmune or inflammatory myopathies. Other terms for HLA class ABC are HLA class I and major histocompatibility complex (MHC) class I.
Immunohistochemistry for the membrane attack complex of complement, C5b-9, also known as MAC, can demontrate capillary expression of this molecule, which can occur in some immune-mediated (inflammatory) myopathies, most notably dermatomyositis. Capillary expression of MAC can be found in diabetic microangiopathy.
Some additional special stains that can be performed on the frozen sample when warranted by the clinical history and findings in the initial battery of stains include the following:
-
For muscular dystrophies, immunohistochemical studies for dystrophin, sarcoglycans, laminin 2-alpha (merosin), and other structural proteins can be performed; the results then can be used to direct special biochemical or genetic analysis that will lead to a specific diagnosis.
-
For some metabolic disorders, studies of the enzymatic activities of myophosphorylase, phosphofructokinase and myoadenylate deaminase can be performed by some laboratories.
-
A stain for acid phosphatase, a lysosomal enzyme, can be useful for the evaluation of certain metabolic disorders and some toxic disorders, as well as in some other circumstances; immunohistochemistry for p62 and LC3, which can be performed on cryostat sections or on formalin fixed paraffin embedded tissue, can also perform this function.
-
For dermatomyositis and certain other disorders, immunofluorescence or immunohistochemistry can be performed to look for the membrane attack complex of complement in blood vessel walls, as noted above, and sarcoplasmic expression of myxovirus resistance A (MxA) can be demonstrated by immunohistochemistry in myofibers in dermatomyositis.
-
For all immune-mediated (inflammatory) myopathies, immunohistology for MHC class I (ie, HLA class ABC or HLA class I) can be performed, if this is not already part of the routine panel of studies.
-
Alpha naphthyl acetate esterase enzyme histochemistry stain can be used to help identify denervated myofibers.
-
Enzyme histochemistry for acid phosphatase can be helpful for diagnosis of lysosomal disorders, both genetic and acquired.
Paraffin sections are usually stained with H&E. The paraffin specimen typically consists of a fairly large surface of myofibers (muscle cells) oriented in the longitudinal direction and a piece in cross-section. A relatively large amount of tissue is usually exposed in each paraffin section; therefore, this specimen is extremely useful for evaluating for processes with a nonuniform distribution (eg, inflammatory myopathies, vasculitis).
Because the fixed, paraffin-embedded specimen exhibits more cytologic detail than seen in the frozen specimen, it is the preferred sample for detecting subtle evidence of myofiber necrosis, determining the type of inflammatory infiltrate present, and examining the structure of blood-vessel walls, the latter particularly in cases of suspected vasculitis.
When indicated, special stains can be performed on the paraffin specimen, including (but not limited to) the following:
-
Special stains for microorganisms, such as bacteria, fungi, and parasites
-
Elastic stains to evaluate for disruption of the elastic lamina of arteries in vasculitis
-
Immunohistochemical stains to determine the subtypes of inflammatory cells within an infiltrate and a variety of other purposes
-
In-situ hybridization for identification of viruses
-
Congo red, thioflavin S or other stains for amyloid
Although a small sample of every muscle biopsy should be set aside for possible EM, performing EM on muscle biopsy samples is not typically a routine procedure; rather, it is usually reserved for selected circumstances in which the pathologist determines that EM has the potential of contributing significantly to determining a specific diagnosis. The pathologist uses knowledge of the clinical history and findings of light-microscopic studies to decide if EM is indicated.
EM is costly and time-consuming and requires a specialized laboratory and technical expertise. Some technical aspects of EM are described below.
Technical aspects of electron microscopy
If the specimen is initially fixed in paraformaldehyde, it is transferred to 3% glutaraldehyde after sufficient time has passed for the paraformaldehyde to penetrate the tissue. The length of time required for this process depends upon the size of the specimen, but overnight fixation is more than satisfactory for the purpose. Glutaraldehyde provides more cross-linking of the membranes, which is needed for EM. There are other fixatives that are appropriate for tissue that will be used for electron microscopy.
If paraformaldehyde is not available, the tissue, held at rest length by pinning to cork, can be placed directly in glutaraldehyde. Because glutaraldehyde does not penetrate the tissue as well as paraformaldehyde does, a specimen placed in glutaraldehyde must be small, approximately 1-2 mm in width and depth. Glutaraldehyde makes tissue brittle and interferes with immunohistochemical studies, so it is not an appropriate fixative for the paraffin specimen.
If the tissue is fixed in formalin, it is not as well preserved for EM as it would be with paraformaldehyde or glutaraldehyde. Performing EM on tissue fixed only in formalin is possible, but this is suboptimal. Cutting tissue out of a paraffin block or removing it from a glass slide for EM is possible, but the likelihood of obtaining useful results with these methods is limited.
After fixation, the tissue is divided into samples of 1 mm3, postfixed with osmium tetroxide, and embedded in epoxy resin. Samples are oriented in either a longitudinal or a transverse direction before polymerization of the resin. The process of embedment for EM requires 2 days.
Survey sections for light microscopy with a thickness of 1 μm, termed semithin (also known as "thick") sections, are cut from the material embedded in plastic and stained with either toluidine blue or methylene blue-azure II. The pathologist reviews these sections, and areas of interest are selected for EM.
An ultramicrotome with a diamond knife is used to cut thin sections for ultrastructural study. These are stained with uranyl acetate and lead citrate. They are placed in an electron microscope and examined.
The following are examples of clinical circumstances in which EM is useful:
-
When evidence is being sought to support a diagnosis of dermatomyositis, EM can be used to look for tubuloreticular inclusions (TRIs) in endothelial cells; if the light-microscopic findings are already diagnostic of dermatomyositis, EM is not necessary.
-
EM can be used to identify inclusions found by light microscopy.
-
EM can help characterize stored material found on light microscopy and define its intracellular localization.
-
EM can be used to analyze structural abnormalities found by light microscopy.
-
EM can assist in the diagnosis of mitochondrial myopathy.
-
EM is only rarely indicated for a muscle that is normal at the light-microscopic level; if normal muscle is found with all of the light-microscopic studies, then this is usually what EM will show, only larger.
There are two exceptions to the final guideline in the above list. One relatively common exception occurs when there is a strong clinical suspicion for dermatomyositis, but the light-microscopic findings are normal. If TRIs are found, they can lend some support to this diagnosis, but if the patient has already been treated with steroids, it is unlikely that TRIs will be present.
The myopathy that is caused by chloroquine or hydroxychloroquine toxicity is typically a vacuolar myopathy, in which vacuoles within myofibers are visible by light microscopy and exhibit excess lysosomal activity, seen with the acid phosphatase stain. In some cases of chloroquine or hydroxychloroquine myopathy, there are no vacuoles within myofibers, but EM reveals the presence of curvilinear bodies, a certain type of inclusion. In an individual with myopathy who has a history of hydroxychloroquine (or chloroquine) ingestion, if the biopsy does not demonstrate the typical vacuolar changes, EM should be performed to look for the characteristic curvilinear bodies within myofibers.
Pearls
Know what you are doing before you arrange or perform the biopsy.
Every muscle pathologist has a series of stories about biopsy procedures that were performed improperly. In many of these situations, the samples were salvaged and yielded diagnoses, but on occasion, the specimen was inadequate for the diagnosis under consideration, or some aspect of the procedure was performed so improperly that the biopsy procedure had to be repeated. For a few examples of suboptimal biopsies that underscore the great importance of using proper technique and following established protocols, see the images of electrocautery coagulation and contraction band artifacts in Technical Issues in Muscle Biopsy, above, and see Skeletal Muscle - Structure and Histology.
Occasional situations exist when the biopsy must be repeated for precise diagnosis and no one is at fault. Such situations include the following:
-
A normal biopsy result without pathologic findings in the setting of a high level of clinical suspicion of a disorder that is known to have a patchy distribution, so the pathology might not be found in a given sample, such as polymyositis
-
An atypical presentation of a rare metabolic disorder, which would not ordinarily be suspected before biopsy, necessitating an additional sample for biochemical assay or other special study, if genetic testing on a sample of blood is not appropriate or available for the disorder of concern or cannot be performed for some other reason
Repeat muscle biopsy is also occasionally indicated to evaluate the patient with known inflammatory myopathy who, after improvement with steroid or other immunosuppressive therapy, develops progressive weakness. Biopsy findings can help distinguish between exacerbation or recurrence of the disorder and steroid myopathy or certain other treatment-associated effects.
Unsuitable, suboptimal, or inadequate biopsy specimens can usually be attributed to lack of planning and forethought, a lack for which there is usually no excuse. The single most important task to remember when one is contemplating muscle biopsy is to contact the pathology laboratory in advance for advice on how to proceed. The specimens required and the preferred method of handling the tissue vary among medical centers. Consulting the center that will receive the biopsy sample is essential for learning the exact requirements and the preferred method of handling and shipping the tissue. The muscle technologists and pathologists and, most important, the patients thank you in advance.
Contraindications and Complications
Few true contraindications for muscle biopsy are noted. This procedure is contraindicated in someone with a high risk of intractable bleeding from the procedure or with an obvious infection at the planned biopsy site.
The surgical procedure to obtain a muscle biopsy is relatively simple and poses little risk to the patient in the absence of an underlying bleeding or clotting disorder.
-
Hematoxylin-eosin (H&E) paraffin section of a muscle biopsy sample reveals end-stage muscle. Fibrovascular and adipose tissues have entirely replaced this muscle, which can therefore provide no information about patient's underlying pathologic process.
-
Hematoxylin and eosin (H&E) paraffin section of this muscle biopsy sample reveals end-stage muscle. The structure in center of this image, consisting of cluster of small muscle fibers surrounded by a fibrous capsule, is a muscle spindle; this is the only finding that confirms the identity of this specimen as skeletal muscle.
-
A fresh muscle biopsy specimen is mounted on cork, in this case using gum tragacanth as the mounting medium. The tissue sample is poised above vial of isopentane, which is chilled with liquid nitrogen coolant. The specimen will be frozen by gently immersing it in the isopentane. In this photograph, myofibers are oriented longitudinally in vertical plane. The tissue will be sectioned by using a cryostat microtome to produce cross-sections of skeletal muscle tissue.
-
Specimen of skeletal muscle on a 10-mm Rayport clamp is fixed in paraformaldehyde. The longitudinal axis of fibers is oriented in horizontal plane. (A small stray piece of muscle that is not clamped lies obliquely over the clamped portion of specimen.)
-
Hematoxylin and eosin paraffin section of a region of a skeletal muscle sample that was coagulated by application of electrocautery to tissue during biopsy procedure. This tissue is unrecognizable.
-
This hematoxylin and eosin (H&E) paraffin section shows severe contraction band artifact in the myofibers in this area of muscle biopsy, which resulted from application of electrocautery. This area of sample is not coagulated, as seen in previous image from a different region of this biopsy, see above image, and thus can be recognized as skeletal muscle, but there is severe disruption of architecture of affected myofibers, which can significantly interfere with interpretation of muscle biopsy.








