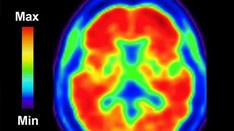
Practice Essentials
Alzheimer disease (AD) is a neurodegenerative disorder marked by cognitive and behavioral impairment that significantly interferes with social and occupational functioning. It is an incurable disease with a long preclinical period and progressive course. In AD, plaques develop in the hippocampus, a structure deep in the brain that helps to encode memories, and in other areas of the cerebral cortex that are involved in thinking and making decisions. Whether plaques themselves cause AD or whether they are a by-product of the AD process remains unknown. The following image depicts one of the cardinal neuroimaging findings in AD – hippocampal atrophy.
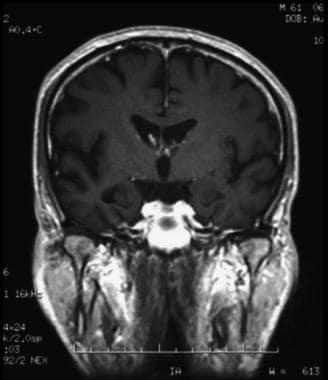 Coronal T1-weighted magnetic resonance imaging (MRI) scan in a patient with moderate Alzheimer disease. Brain image reveals hippocampal atrophy, especially on the right side.
Coronal T1-weighted magnetic resonance imaging (MRI) scan in a patient with moderate Alzheimer disease. Brain image reveals hippocampal atrophy, especially on the right side.
Signs and symptoms
Preclinical Alzheimer disease
A patient with preclinical AD may appear completely normal on physical examination and mental status testing. Specific regions of the brain (eg, entorhinal cortex, hippocampus) are likely to be affected decades before any signs or symptoms appear.
Mild Alzheimer disease
Signs of mild AD can include the following:
-
Memory loss
-
Confusion about the location of familiar places
-
Taking longer to accomplish normal, daily tasks
-
Trouble handling money and paying bills
-
Compromised judgment, often leading to bad decisions
-
Loss of spontaneity and sense of initiative
-
Mood and personality changes; increased anxiety
Moderate Alzheimer disease
The symptoms of this stage can include the following:
-
Increasing memory loss and confusion
-
Shortened attention span
-
Problems recognizing friends and family members
-
Difficulty with language; problems with reading, writing, working with numbers
-
Difficulty organizing thoughts and thinking logically
-
Inability to learn new things or to cope with new or unexpected situations
-
Restlessness, agitation, anxiety, tearfulness, wandering, especially in the late afternoon or at night
-
Repetitive statements or movement; occasional muscle twitches
-
Hallucinations, delusions, suspiciousness or paranoia, irritability
-
Loss of impulse control: Shown through behavior such as undressing at inappropriate times or places or vulgar language
-
Perceptual-motor problems: Such as trouble getting out of a chair or setting the table
Severe Alzheimer disease
Patients with severe AD cannot recognize family or loved ones and cannot communicate effectively. They are completely dependent on others for care, and all sense of self seems to vanish.
Other symptoms of severe AD can include the following:
-
Weight loss
-
Seizures, skin infections, difficulty swallowing
-
Groaning, moaning, or grunting
-
Increased sleeping
-
Lack of bladder and bowel control
In end-stage AD, patients may be in bed much or all of the time. Death is often the result of other illnesses, frequently aspiration pneumonia.
See Clinical Presentation for more detail.
Diagnosis
Means of diagnosing AD include the following:
-
Clinical examination: The clinical diagnosis of AD is usually made during the mild stage of the disease, using the above-listed signs
-
Lumbar puncture: levels of tau and phosphorylated tau in the cerebrospinal fluid are often elevated in AD, whereas amyloid levels are usually low; at present, however, routine measurement of CSF tau and amyloid is not recommended except in research settings
-
Imaging studies: Imaging studies are particularly important for ruling out potentially treatable causes of progressive cognitive decline, such as chronic subdural hematoma or normal-pressure hydrocephalus. [1] Moreover, volumetric studies of the hippocampus and 2-[18F]fluoro-2-Deoxy-D-glucose positron emission tomography (FDG-PET) with or without amyloid imaging have been employed for early detection and differentiating dementia etiologies. [2]
See Clinical Presentation and Workup for more detail.
Management
All drugs approved by the US Food and Drug Administration (FDA) for the treatment of AD are symptomatic therapies that modulate neurotransmitters, either acetylcholine or glutamate. The standard medical treatment for AD includes cholinesterase inhibitors (ChEIs) and a partial N-methyl-D-aspartate (NMDA) antagonist. [3, 4] They do not treat the underlying cause of AD nor halt the rate of decline. Additionally, amyloid-directed antibodies (eg, aducanumab, lecanemab) have been approved.
The following classes of psychotropic medications have been used to treat the secondary symptoms of AD, such as depression, agitation, aggression, hallucinations, delusions, and sleep disorders [5] :
-
Antidepressants
-
Anxiolytics
-
Antiparkinsonian agents
-
Beta-blockers
-
Antiepileptic drugs
-
Neuroleptics
Prevention
There are no proven modalities for preventing AD, [3] but evidence, largely epidemiologic, suggests that healthy lifestyles can reduce the risk of developing the disease; the following may be protective [6, 7] :
-
Physical activity
-
Exercise
-
Cardiorespiratory fitness
-
Diet: Although no definitive dietary recommendations can be made, in general, nutritional patterns that appear beneficial for AD prevention fit the Mediterranean diet
See Treatment and Medication for more detail.
Background
Alzheimer disease (AD) is the most common form of dementia. In the United States alone, approximately 6.08 million Americans had either clinical AD or mild cognitive impairment due to AD in 2017. That number is expected to grow to 15 million by 2060. [8] AD is the sixth leading cause of death in the United States, accounting for 3.6% of all deaths in 2014. [9] The percentage of Alzheimer’s decedents who died in a medical facility (e.g., hospital) declined from 14.7% in 1999 to 6.6% in 2014, whereas the percentage who died at home increased from 13.9% in 1999 to 24.9% in 2014. [9] Economically, AD is a major public health problem. Total payments in 2017 for health care and long-term care for all individuals with AD or other dementias are estimated at $259 billion. By 2050, these costs could rise as high as $1.1 trillion. [10]
Currently, an autopsy or brain biopsy is the only way to make a definitive diagnosis of AD. In clinical practice, the diagnosis is usually made on the basis of the history and findings on Mental Status Examination (see Presentation).
Symptomatic therapies are the only treatments available for AD. The standard medical treatments include cholinesterase inhibitors and a partial N -methyl-D-aspartate (NMDA) antagonist. Psychotropic medications are often used to treat secondary symptoms of AD, such as depression, agitation, and sleep disorders. (See Treatment.)
For additional information, see Alzheimer’s Disease: Slideshow.
Historical background
In 1901, a German psychiatrist named Alois Alzheimer observed a patient at the Frankfurt Asylum named Mrs. Auguste D. This 51-year-old woman suffered from a loss of short-term memory, among other behavioral symptoms that puzzled Dr. Alzheimer. Five years later, in April 1906, the patient died, and Dr. Alzheimer sent her brain and her medical records to Munich, where he was working in the lab of Dr. Emil Kraeplin. By staining sections of her brain in the laboratory, he was able to identify amyloid plaques and neurofibrillary tangles. [11]
A speech given by Dr. Alzheimer on November 3, 1906, was the first time the pathology and the clinical symptoms of the disorder, which at the time was termed presenile dementia, were presented together. Alzheimer published his findings in 1907. [12]
In the past 15-20 years, dramatic progress has been made in understanding the neurogenetics and pathophysiology of AD (see Pathophysiology and Etiology). Four different genes have been definitively associated with AD, and others that have a probable role have been identified. The mechanisms by which altered amyloid and tau protein metabolism, inflammation, oxidative stress, and hormonal changes may produce neuronal degeneration in AD are being elucidated, and rational pharmacologic interventions based on these discoveries are being developed.
Anatomy
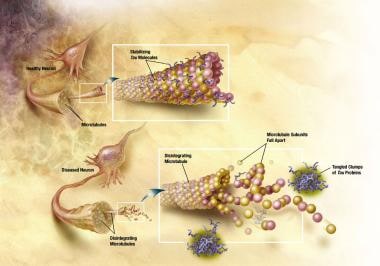
Healthy neurons have an internal support structure partly made up of structures called microtubules. These microtubules act like tracks, guiding nutrients and molecules from the body of the cell down to the ends of the axon and back. A special kind of protein, tau, binds to the microtubules and stabilizes them.
In AD, tau is changed chemically. It begins to pair with other threads of tau, which become tangled together. When this happens, the microtubules disintegrate, collapsing the neuron’s transport system (see the image below). The formation of these neurofibrillary tangles (NFTs) may result first in malfunctions in communication between neurons and later in the death of the cells.
In addition to NFTs, the anatomic pathology of AD includes senile plaques (SPs; also known as beta-amyloid plaques) at the microscopic level and cerebrocortical atrophy at the macroscopic level (see the image below). The hippocampus and medial temporal lobe are the initial sites of tangle deposition and atrophy. [13] This can be seen on brain magnetic resonance imaging early in AD and helps support a clinical diagnosis.
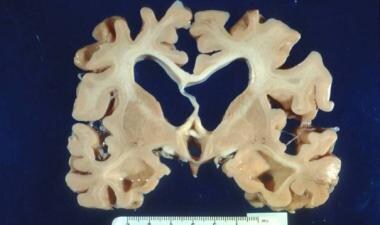
SPs and NFTs were described by Alois Alzheimer in his original report on the disorder in 1907. [12] They are now universally accepted as the pathological hallmark of the disease.
Pathophysiology
A continuum exists between the pathophysiology of normal aging and that of AD. [14] Pathologic hallmarks of AD have been identified; however, these features also occur in the brains of cognitively intact persons. For example, in a study in which neuropathologists were blinded to clinical data, they identified 76% of brains of cognitively intact elderly patients as demonstrating AD. [15]
AD affects the 3 processes that keep neurons healthy: communication, metabolism, and repair. Certain nerve cells in the brain stop working, lose connections with other nerve cells, and finally die. The destruction and death of these nerve cells causes the memory failure, personality changes, problems in carrying out daily activities, and other features of the disease.
The accumulation of SPs primarily precedes the clinical onset of AD. NFTs, loss of neurons, and loss of synapses accompany the progression of cognitive decline. [14]
Considerable attention has been devoted to elucidating the composition of SPs and NFTs to find clues about the molecular pathogenesis and biochemistry of AD. The main constituent of NFTs is the microtubule-associated protein tau (see Anatomy). In AD, hyperphosphorylated tau accumulates in the perikarya of large and medium pyramidal neurons. Somewhat surprisingly, mutations of the tau gene result not in AD but in some familial cases of frontotemporal dementia.
Since the time of Alois Alzheimer, SPs have been known to include a starchlike (or amyloid) substance, usually in the center of these lesions. The amyloid substance is surrounded by a halo or layer of degenerating (dystrophic) neurites and reactive glia (both astrocytes and microglia).
One of the most important advances in recent decades has been the chemical characterization of this amyloid protein, the sequencing of its amino acid chain, and the cloning of the gene encoding its precursor protein (on chromosome 21). These advances have provided a wealth of information about the mechanisms underlying amyloid deposition in the brain, including information about the familial forms of AD. (See Amyloid Hypothesis Versus Tau Hypothesis, below.)
Alzheimer disease biomarkers may follow a sequential pattern in the brain, according to a study of AD biomarker trajectories. The study included both symptomatic and asymptomatic carriers of autosomal dominant gene mutations linked to AD, including APP, PSEN1, and PSEN2. Researchers did not address tauopathy. Results show that amyloid deposition in the brain occurs first, followed by a decline in glucose metabolism and then structural brain atrophy. The rate of Ab accumulation was significantly higher in mutation carriers compared to noncarriers, and was found to begin more than 2 decades before the expected onset of dementia. In carriers, metabolism began to decrease at a mean of 14.1 years before expected symptom onset, and structural changes in the brain began 4.7 years before expected symptom onset. It is important to note that only about 1% of patients with AD have an autosomal dominant mutation, so results may not be generalizable to sporadic AD. [16]
Although the amyloid cascade hypothesis has gathered the most research financing, other interesting hypotheses have been proposed. Among these are the mitochondrial cascade hypothesis. [17]
In addition to NFTs and SPs, many other lesions of AD have been recognized since Alzheimer’s original papers were published. These include the granulovacuolar degeneration of Shimkowicz; the neuropil threads of Braak et al [17] ; and neuronal loss and synaptic degeneration, which are thought to ultimately mediate the cognitive and behavioral manifestations of the disorder.
In 2019, researchers identified a new type of dementia that mimics AD but that is caused by another mechanism in the brain. This new classification is limbic-predominant age-related TDP-43 encephalopathy, (LATE). TDP-43 is a protein that helps regulate gene expression in the brain and other tissues, and when it misfolds it causes problems in the brain. According to researchers, misfolded TDP-43 protein is very common in older adults; about 25% of people aged 85 and older have enough misfolded TDP-43 protein to affect their memory and thinking skills. [18]
Neurofibrillary tangles and senile plaques
Plaques are dense, mostly insoluble deposits of protein and cellular material outside and around the neurons. Plaques are made of beta-amyloid (Ab), a protein fragment snipped from a larger protein called amyloid precursor protein (APP). These fragments clump together and are mixed with other molecules, neurons, and non-nerve cells (see the images below).
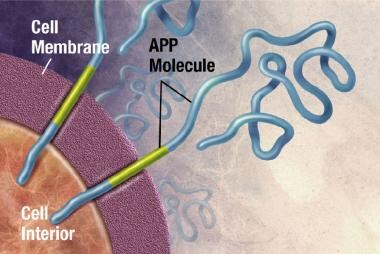 APP is associated with the cell membrane, the thin barrier that encloses the cell. After it is made, APP sticks through the neuron's membrane, partly inside and partly outside the cell. Image courtesy of NIH.
APP is associated with the cell membrane, the thin barrier that encloses the cell. After it is made, APP sticks through the neuron's membrane, partly inside and partly outside the cell. Image courtesy of NIH.
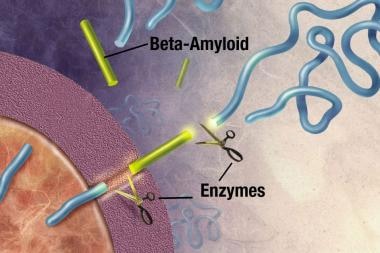 Enzymes (substances that cause or speed up a chemical reaction) act on the APP and cut it into fragments of protein, one of which is called beta-amyloid. Image courtesy of NIH.
Enzymes (substances that cause or speed up a chemical reaction) act on the APP and cut it into fragments of protein, one of which is called beta-amyloid. Image courtesy of NIH.
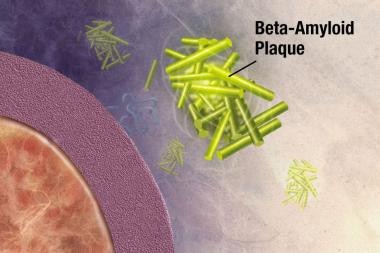 The beta-amyloid fragments begin coming together into clumps outside the cell, then join other molecules and non-nerve cells to form insoluble plaques. Image courtesy of NIH.
The beta-amyloid fragments begin coming together into clumps outside the cell, then join other molecules and non-nerve cells to form insoluble plaques. Image courtesy of NIH.
In AD, plaques develop in the hippocampus, a structure deep in the brain that helps to encode memories, and in other areas of the cerebral cortex that are used in thinking and making decisions. Plaques may begin to develop as early as the fifth decade of life. [19] Whether Ab plaques themselves cause AD or whether they are a by-product of the AD process is still unknown. It is known that changes in APP structure can cause a rare, inherited form of AD.
Tangles are insoluble twisted fibers that build up inside the nerve cell. Although many older people develop some plaques and tangles, the brains of people with AD have them to a greater extent, especially in certain regions of the brain that are important in memory. There are likely to be significant age-related differences in the extent to which the presence of plaques and tangles are indicative of the presence of dementia.
NFTs are initially and most densely distributed in the medial aspect and in the pole of the temporal lobe; they affect the entorhinal cortex and the hippocampus most severely (however, Braak et al found that in sporadic AD, tauopathy may appear first in the lower brainstem rather than in the transentorhinal region [19] ). As AD progresses, NFTs accumulate in many other cortical regions, beginning in high-order association regions and less frequently in the primary motor and sensory regions.
SPs also accumulate primarily in association cortices and in the hippocampus. Plaques and tangles have relatively discrete and stereotypical patterns of laminar distribution in the cerebral cortex, which indicate predominant involvement of corticocortical connections.
Although NFTs and SPs are characteristic of AD, they are not pathognomonic. NFTs are found in several other neurodegenerative disorders, including progressive supranuclear palsy and dementia pugilistica (chronic traumatic encephalopathy). SPs may occur in normal aging.
Therefore, the mere presence of these lesions is not sufficient to support the diagnosis of AD. These lesions must be present in sufficient numbers and in a characteristic topographic distribution to fulfill the current histopathologic criteria for AD. There is consensus that the presence of even low numbers of NFTs in the cerebral neocortex with concomitant SPs is characteristic of AD.
Some authorities believed that NFTs, when present in low densities and essentially confined to the hippocampus, were part of normal aging. However, the histologic stages for AD that Braak et al formulated include an early stage in which NFTs are present at a low density in the entorhinal and perirhinal (ie, transentorhinal) cortices. [20] Therefore, even small numbers of NFTs in these areas of the medial temporal lobe may be abnormal.
Amyloid hypothesis versus tau hypothesis
A central but controversial issue in the pathogenesis of AD is the relationship between amyloid deposition and NFT formation. Evidence shows that abnormal amyloid metabolism plays a key pathogenic role. At high concentrations, the fibrillar form of Ab has been shown to be neurotoxic to cultured neurons.
Cultured cortical and hippocampal neurons treated with Ab protein exhibit changes characteristic of apoptosis (self-regulated cell destruction), including nuclear chromatin condensation, plasma membrane blebbing, and internucleosomal DNA fragmentation. The fibrillar form of Ab has also been shown to alter the phosphorylation state of tau protein.
The identification of several point mutations within the APP gene in some patients with early-onset familial AD and the development of transgenic mice exhibiting cognitive changes and SPs also incriminate Ab in AD. The apolipoprotein E (APOE) E4 allele, which has been linked with significantly increased risk for developing AD, may promote inability to suppress production of amyloid, increased production of amyloid, or impaired clearance of amyloid with collection outside of the neuron.
Autopsies have shown that patients with 1 or 2 copies of the APOE E4 allele tend to have more amyloid. Additional evidence comes from recent experimental data supporting the role of presenilins in Ab metabolism, as well as findings of abnormal production of Ab protein in presenilin-mutation familial Alzheimer disease.
Although very popular, the amyloid hypothesis is not uniformly accepted. On post-mortem analysis, amyloid plaques may be undetectable in the brains of patients who had severe AD but may be present in the brains of elderly patients who did not have dementia. [21]
Dementia severity correlates better with the number of neocortical NFTs than with SPs. The tau protein stabilizes neuronal microtubules. Destabilization of the microtubular system is speculated to disrupt the Golgi apparatus, in turn inducing abnormal protein processing and increasing production of Ab. In addition, this destabilization may decrease axoplasmic flow, generating dystrophic neurites and contributing to synaptic loss.
For more information, see the Medscape Reference article Alzheimer Disease and APOE-4.
Granulovacuolar degeneration and neuropil threads
Granulovacuolar degeneration occurs almost exclusively in the hippocampus. Neuropil threads are an array of dystrophic neurites diffusely distributed in the cortical neuropil, more or less independently of plaques and tangles. This lesion suggests neuropil alterations beyond those merely due to NFTs and SPs and indicates an even more widespread insult to the cortical circuitry than that visualized by studying only plaques and tangles.
Cholinergic neurotransmission and Alzheimer disease
The cholinergic system is involved in memory function, and cholinergic deficiency has been implicated in the cognitive decline and behavioral changes of AD. Activity of the synthetic enzyme choline acetyltransferase (CAT) and the catabolic enzyme acetylcholinesterase are significantly reduced in the cerebral cortex, hippocampus, and amygdala in patients with AD.
The nucleus basalis of Meynert and diagonal band of Broca provide the main cholinergic input to the hippocampus, amygdala, and neocortex, which are lost in patients with AD. Loss of cortical CAT and decline in acetylcholine synthesis in biopsy specimens have been found to correlate with cognitive impairment and reaction-time performance. Because cholinergic dysfunction may contribute to the symptoms of patients with AD, enhancing cholinergic neurotransmission constitutes a rational basis for symptomatic treatment.
Oxidative stress and damage
Oxidative damage occurs in AD. Studies have demonstrated that an increase in oxidative damage selectively occurs within the brain regions involved in regulating cognitive performance. [22]
Oxidative damage potentially serves as an early event that then initiates the development of cognitive disturbances and pathological features observed in AD. A decline in protein synthesis capabilities occurs in the same brain regions that exhibit increased levels of oxidative damage in patients with mild cognitive impairment (MCI) and AD. Protein synthesis may be one of the earliest cellular processes disrupted by oxidative damage in AD. [23]
Oxidative stress is believed to be a critical factor in normal aging and in neurodegenerative diseases such as Parkinson disease, amyotrophic lateral sclerosis, and AD. Formation of free carbonyls and thiobarbituric acid-reactive products, an index of oxidative damage, are significantly increased in AD brain tissue compared with age-matched controls. Plaques and tangles display immunoreactivity to antioxidant enzymes.
Multiple mechanisms exist by which cellular alterations may be induced by oxidative stress, including production of reactive oxygen species (ROS) in the cell membrane (lipid peroxidation). This in turn impairs the various membrane proteins involved in ion homeostasis such as N -methyl-D-aspartate receptor channels or ion-motive adenosine triphosphatases.
The subsequent increase in intracellular calcium, along with the accumulation of ROS, damages various cellular components such as proteins, DNA, and lipids and may result in apoptotic cellular death. Increased intracellular calcium may also alter calcium-dependent enzyme activity such as the implication of protein kinase C in amyloid protein metabolism and the phosphorylation of tau.
The involvement of calcium in AD has suggested that blocking the increase in free intracellular calcium may diminish neuronal injury. However, clinical trials of nimodipine, a lipophilic calcium channel blocker that is mediated through inactivation of voltage-dependent L-type (long-lasting) calcium channels, have yielded generally disappointing results in patients with AD.
The apoptotic pattern of cellular death seen in oxidative stress is similar to that produced by Ab peptide exposure, and Ab neurotoxicity is attenuated by antioxidants such as vitamin E. Ab may induce toxicity by engaging several binding sites on the membrane surface. The receptor for advanced glycation end products (RAGE) may be one of these receptors. RAGE is a member of the immunoglobulin superfamily of cell surface molecules known for its capacity to bind advanced glycation end products.
RAGE is also expressed in a variety of other cell types, including endothelial cells and mononuclear phagocytes. Activation of this receptor is believed to trigger cellular oxidative reactions. In addition, RAGE has been shown to mediate the interaction of Ab with glial cells, which may be one of the first steps in the inflammatory cascade.
Inflammatory reactions
Inflammatory and immune mechanisms may play a role in the degenerative process in AD. Reactive microglia are embedded in neuritic plaques. Increased cytokine levels are seen in the serum, cortical plaques, and neurons of patients with AD, as compared with aged-matched control patients. Interestingly, transforming growth factor beta 1 (TGF-β1), which is an anti-inflammatory cytokine, has been found to promote or accelerate the deposition of amyloid.
Classical complement pathway fragments are also found in the brains of patients with AD, and amyloid may directly activate the classical complement pathway in an antibody-independent fashion.
Whether markers of immune and inflammatory processes actively participate in the neurodegenerative process or instead represent an epiphenomenon remains unclear. Brain specimens from elderly patients with arthritis treated with nonsteroidal anti-inflammatory drugs (NSAIDs) have similar numbers of senile plaques as do control brains.
However, less microglial activation is seen in the brains of the patients with arthritis. This suggests that although NSAIDs may not impede senile plaque formation, they may delay or prevent clinical symptoms by limiting the associated inflammation.
As mentioned above, RAGE has been shown to mediate the interaction of amyloid and glial cells, producing cellular activation and an inflammatory response with cytokine production, chemotaxis, and haptotaxis. The expression of this receptor appears to be upregulated in neurons, vasculature, and microglia in affected regions of AD brains.
The unrelated class A scavenger receptor (class A SR) also mediates the adhesion of microglial cells to amyloid fibrils. SPs contain high concentrations of microglia that express class A SRs. RAGE and class A SRs may represent novel pharmacologic targets for diminishing the inflammatory and oxidative reactions associated with AD.
Clusterin
Clusterin, a plasma protein, plays an important role in the pathogenesis of AD. In one study, clusterin was associated with atrophy of the entorhinal cortex, baseline disease severity, and rapid clinical progression in AD. This important study suggests that alterations in amyloid chaperone proteins could be a relevant peripheral signature of AD. [24] A study by Schrijvers et al notes that although plasma clusterin levels are significantly associated with baseline prevalence and severity of AD, they are not related to the risk for AD. [25]
Presenilins
A significant proportion of early-onset autosomal-dominant AD cases have been linked to a candidate gene on chromosome 14 (14q24.3) called presenilin-1 (PS1) and a candidate gene on chromosome 1 called presenilin-2 (PS2). The 2 putative products of these candidate genes, PS1 and PS2, share substantial amino-acid and structural similarities, suggesting that they may be functionally related. In addition, the expression patterns of PS1 and PS2 in the brain are similar, if not identical.
Both PS1 messenger RNA (mRNA) and PS2 mRNA are detectable only within neuronal populations. Immunochemical analyses indicate that PS1 localizes to intracellular compartments, such as the endoplasmic reticulum and the Golgi complex, that are involved in similar functions. Evidence supports the role of presenilins in Ab metabolism. Mice deficient in the expression of PS1 exhibit a dramatic decrease in proteolytic cleavage of the transmembrane domain of amyloid precursor protein (APP) by secretase.
PS1 is immunoreactive with the neuritic component of SPs. Both asymptomatic and demented subjects carrying the PS1 mutation have increased production of the amyloidogenic Ab 42/43 isoform in skin fibroblasts and plasma. Prominent deposition of Ab 42/43 is found in many brain regions of patients with PS1 mutations. These findings, in suggesting that inhibiting presenilin function might decrease Ab amyloid production, offer new therapeutic avenues.
Estrogen loss
Postmenopausal women are at higher risk than men for AD. Some studies have shown that estrogen loss may lead to cognitive decline and neuronal degeneration, and the expression of nerve growth factor and brain-derived neurotrophic factor mRNA is also decreased.
Estrogen has also been shown to exert cytoprotective effects and to prevent amyloid toxicity in human neuroblastoma cell cultures. However, a randomized clinical trial of estrogen in cognitively normal women aged 65 years and older with a first-degree relative with AD showed that estrogen therapy might actually increase the risk of stroke and dementia. [26]
Etiology
The cause of AD is unknown. Several investigators now believe that converging environmental and genetic risk factors trigger a pathophysiologic cascade that, over decades, leads to Alzheimer pathology and dementia. [27]
The following risk factors for Alzheimer-type dementia have been identified: [28, 29, 30, 31]
-
Advancing age
-
Family history
-
APOE 4 genotype
-
Obesity
-
Insulin resistance
-
Vascular factors
-
Dyslipidemia
-
Hypertension
-
Inflammatory markers
-
Down syndrome
-
Traumatic brain injury
Midlife hypertension is an established risk factor for late-life dementia, of which AD is the most common type. A brain autopsy study evaluating the link between hypertension and AD found that patients using beta-blockers to control blood pressure had fewer Alzheimer's-type brain lesions on autopsy compared to patients taking no drug therapy or those taking other medications. [32]
In addition, epidemiologic studies have suggested some possible risk factors such as aluminum [33, 34] and previous depression [35] . Other studies have suggested protective factors (e.g.,education [36, 37] , long-term use of nonsteroidal anti-inflammatory drugs [38] ).
Genetic causes
Although most cases of AD are sporadic (ie, not inherited), familial forms of AD do exist. Autosomal dominant AD, which accounts for less than 5% of cases, is almost exclusively early onset AD; cases occur in at least 3 individuals in 2 or more generations, with 2 of the individuals being first-degree relatives. [39]
Familial clustering represents approximately 15–25% of late-onset AD cases and most often involves late-onset AD. In familial clustering, at least 2 of the affected individuals are third-degree relatives or closer. [39]
Mutations in the following genes unequivocally cause early-onset autosomal dominant AD:
-
The amyloid precursor protein (APP) gene on chromosome 21
-
The presenilin-1 (PS1) gene on chromosome 14
-
The presenilin-2 (PS2) gene on chromosome 1
All 3 of these genes lead to a relative excess in the production of the stickier 42-amino acid form of the Ab peptide over the less sticky 40-amino-acid form.
This beta-pleated peptide is postulated to have neurotoxic properties and to lead to a cascade of events (as yet incompletely understood) that results in neuronal death, synapse loss, and the formation of NFTs and SPs, among other lesions. Nonetheless, the mutations that have been found to date account for less than half of all cases of early-onset AD.
Other than the apolipoprotein E epsilon 4 (APOE E4) genotype, no polymorphisms in other genes have been consistently found to be associated with late-onset AD. However, genome-wide association studies have identified the following additional susceptibility loci [40] :
-
Clusterin (CLU) gene
-
Phosphatidylinositol-binding clathrin assembly protein (PICALM) gene
-
Complement receptor 1 (CR1) gene
-
ATP-binding cassette sub-family A member 7 gene (ABCA7)
-
Membrane-spanning gene cluster (MS4A6A/MS4A4E)
-
Ephrin receptor A1 (EPHA1)
-
CD33
-
CD2AP
APP mutations
The observation that patients with Down syndrome (trisomy 21) develop cognitive deterioration and typical pathological features of AD by middle age led to the discovery of the APP gene on chromosome 21. Simultaneously, a locus segregating with a minority of early-onset familial AD kindreds was mapped to this chromosome, in the same region as the APP gene. For more information, see the Medscape Reference article Alzheimer Disease in Down Syndrome.
Subsequently, several missense mutations within the APP gene that resulted in amino acid substitutions in APP were identified in these familial AD kindreds. Such mutations appear to alter the previously described proteolytic processing of APP, generating amyloidogenic forms of Ab.
Skin fibroblasts from individuals carrying APP mutations produce increased Ab 42/43. Increased plasma concentration of Ab 42/43 is also seen in these patients, regardless of age, sex, or clinical status. Interestingly, some patients with sporadic AD may exhibit similar elevations of plasma Ab 42/43.
PS1 and PS2 mutations
Approximately 50-70% of early-onset autosomal-dominant AD cases appear to be associated with a locus (AD3) mapped by genetic linkage to the long arm of chromosome 14 (14q24.3). Numerous missense mutations have been identified on a strong candidate gene, called PS1.
At the same time, another autosomal dominant locus responsible for early-onset AD was localized to chromosome 1. Two mutations were identified on the candidate gene, designated PS2. The physiological role of presenilins and the pathogenic effects of their mutations are not yet well understood.
APOE
The gene encoding the cholesterol-carrying apolipoprotein E (APOE) on chromosome 19 has been linked to increased risk for AD, principally late-onset but also some early-onset cases. The gene is inherited as an autosomal codominant trait with 3 alleles. The APOE E2 allele, the least prevalent of the 3 common APOE alleles, is associated with the lowest risk of developing AD, [41] with a lower rate of annual hippocampal atrophy and higher cerebrospinal fluid Aβ and lower phosphotau, suggesting less AD pathology. [42]
The E3 allele confers intermediate risk of developing AD, with less risk than the E4 allele. The E3 allele, which is more common than the E2 allele, may protect tau from hyperphosphorylation, and the E2 allele’s effect on tau phosphorylation is complex.
APOE E4 gene “dose” is correlated with increased risk and earlier onset of AD. [43] Individuals who are genetically predisposed to AD are advised to closely control their blood pressure closely. Hypertension has been shown to interact with APOE E4 genotype to increase amyloid deposition in cognitively healthy middle-aged and older adults; controlling hypertension may significantly decrease the risk of developing amyloid deposits, even in those with genetic risk. [44, 45]
Persons with 2 copies of the APOE E4 allele (4/4 genotype) have a significantly greater risk of developing AD than persons with other APOE subtypes. Mean age at onset is significantly lower in the presence of 2 APOE E4 copies. A collaborative study has suggested that APOE E4 exerts its maximal effect before the age of 70 years.
Many APOE E4 carriers do not develop AD, and many patients with AD do not have this allele. Therefore, the presence of an APOE E4 allele does not secure the diagnosis of AD, but instead, the APOE E4 allele acts as a biologic risk factor for the disease, especially in those younger than 70 years.
According to one study, exposure to particulate matter (PM) in the ambient air and its interactions with APOE alleles may contribute to the acceleration of brain aging and the pathogenesis of AD. In addition to examining experimental mouse models, researchers analyzed data on more than 3,600 US women between the ages of 65 and 79 years from 48 states. None of the women had dementia when the study began. Data show that residing in places with fine PM exceeding EPA standards increased the risks for global cognitive decline and all-cause dementia respectively by 81 and 92%, with stronger adverse effects in APOE ε4/4 carriers. These joint data from humans and mice provide the first evidence that neurodegenerative effects of airborne PM may involve gene-environment interactions with APOE ε4. [46]
Insulin resistance
A small study by Baker et al implies that insulin resistance, as evidenced by decreased cerebral glucose metabolic rate measured by a specific type of positron emission tomography (PET) scan, may be useful as an early marker of AD risk, even before the onset of MCI. [47] The PET scan revealed a qualitatively different activation pattern in patients with prediabetes or type 2 diabetes mellitus during a memory encoding task, as compared with healthy individuals who were not insulin resistant.
Although the study by Baker et al had too few subjects (n=23) for the results to reach statistical significance, a study by Schrijvers et al in a much larger population (3,139 subjects) found a similar association between insulin resistance and AD over 3 years, which then disappeared after that time. [48] These researchers used a different measure of insulin resistance, the homeostasis model assessment. Disturbances in insulin metabolism may not cause neurological changes but may influence and accelerate these changes, leading to an earlier onset of AD.
Infection
An emerging field of research suggests a significant association between AD and chronic infection with various species of spirochetes, including the periodontal pathogen Treponemas and Borrelia burgdorferi, as well as pathogens such as herpes simplex virus type 1. [49] In vitro and animal studies support the concept of infection resulting in chronic inflammation and neuronal destruction. Ab has been shown to be an antimicrobial peptide, so its accumulation might represent a response to infection.
Depression
Depression has been identified as a risk factor for AD and other dementias. Recent Framingham data have helped bolster the epidemiological association. The study showed a 50% increase in AD and dementia in those who were depressed at baseline. [50] During a 17-year follow-up period, a total of 21.6% of participants who were depressed at baseline developed dementia, as compared with 16.6% of those who were not depressed.
In another related study, recurrent depression was noted to be particularly pernicious. One episode of depression conferred an 87–92% increase in dementia risk, while having 2 or more episodes nearly doubled the risk. [51]
According to the results of a meta-analysis of 23 population-based, prospective cohort studies, late-life depression is associated with an increased risk for all-cause dementia, vascular dementia, and AD. [52, 53] The risk for vascular dementia appeared to be significantly higher than the risk for AD. The analysis included data on patients 50 years and older who were free of dementia at baseline. The total sample included in the pooled analysis for all-cause dementia was 49,612 participants, 5116 of whom had late-life depression.
Alternatively, a large, longitudinal study found that depression that starts early in life increases the risk for AD. Researchers used data from the Prospective Population Study of Women in Gothenburg Sweden, which began in 1968. The study sample included 800 women (mean age, 46 years), born between 1914 and 1930, who were followed up with in 1974, 1980, 1992, 2000, 2009, and 2012. Data show those women who experienced the onset of depression before age 20 years were three times more likely to develop AD (adjusted HR, 3.41; 95% CI, 1.78 - 6.54). [35]
Head trauma
Moderate to severe head trauma has been documented as a risk factor for the development of AD as well as other forms of dementia later in life. [54] Chen et al have proposed that traumatic brain injury leads to accumulation of amyloid precursor protein with its proteolytic enzymes at sites of axonal injury, increased intracellular production of Ab, release of Ab from injured axons into the extracellular space, and deposition of Ab into extracellular plaques. [55]
A study that followed over 7,000 US veterans of World War II showed that those who had sustained head injuries had twice the risk of developing dementia later in life, with veterans who suffered more severe head trauma being at an even higher risk. The study also found that the presence of the APOE gene and sustaining head trauma seemed to act additively to increase the risk of developing AD, although there was no direct correlation. [56]
Epigenetics
Epigenetics is a change in gene expression that results from gene-environment interactions. This is mediated by DNA methylation, RNA editing, and RNA interference without changes in the DNA sequence. Epigenetic elements in AD are suggested by facts that the majority of cases of AD are sporadic, occur in patients without a family history of the disease, and have onset late in life.
One environmental factor that has shown damage in laboratory animals consistent with human AD is lead. Early exposure to lead in monkeys resulted in plaque formation as they aged. [57] One aspect of early lead exposure appears to be increased oxidative stress in brain cells. Oxidative stress is the accumulation of excess free radicals that alter methylation patterns in the cells.
Early oxidative stress other than lead has been postulated as one cause of sporadic AD. Brain cells in AD exhibit overexpression and repression of AD genes, suggesting hypomethylation and hypermethylation, which are associated with oxidative stress. [58]
Given that lead exposure to animals in early life does not produce manifestations until later in life, continued environmental stress may contribute to expression. Consequently, it is possible that the use of antioxidant supplements starting in childhood might decrease long-term oxidative stress and decrease the incidence of AD. The work of Harman indicates that antioxidants may decrease cell damage and aging by decreasing excess oxidative stress. [59]
The only major study of one antioxidant, vitamin E, yielded disappointing results. However, the study involved a very limited time usage. At present no other changes in environmental exposures have been studied for prevention of AD, but this area will be critical in future long-term studies.
Epidemiology
United States statistics
According to a 2017 report, AD affects an estimated 6.08 million people in the United States, and approximately 200,000 people younger than 65 years with AD constitute the younger-onset US population with AD. [10, 8] A larger number of individuals have decreased cognitive function (eg, mild cognitive impairment); this condition frequently evolves into full-blown dementia, thereby increasing the number of affected persons. By the year 2050, AD could affect 13.8 million persons in the United States. [10]
Using data from Rochester, Minnesota, Genin et al calculated that at the age of 85 years, the lifetime risk of AD without reference to APOE genotype was 11% in males and 14% in females; for APOE 4/4 carriers, risk ranged from 51% for males to 60% for females. For APOE 3/4 carriers, risk ranged from 23% for males to 30% for females. In French incidence data, lifetime risk for females at age 85 was 68% for APOE 4/4 carriers and 35% for APOE 3/4 carriers. [60]
In the United States, AD is a leading cause of death. While deaths from other major causes have been decreasing, deaths from of AD have been rising. [10] AD was the sixth leading cause of death in 2017. [10, 61, 62] Moreover, AD as an underlying cause of death is frequently underreported. [63]
International statistics
Prevalence rates of AD similar to those in the United States have been reported in industrialized nations. The prevalence of dementia in persons 65 years of age and older in North America is approximately 6-10%, with AD accounting for two-thirds of these cases. If milder cases are included, the prevalence rates double. Countries experiencing rapid increases in the elderly segments of their population have rates approaching those in the United States.
The World Health Organization’s review in 2000 on the Global Burden of Dementia, [64] which was an integrative analysis of 47 surveys across 17 countries, suggested that approximate rates for dementia from any cause are under 1% in persons aged 60-69 years, rising to about 39% in persons 90-95 years old. The prevalence doubles with every 5 years of age within that range, with few differences taking into account secular changes, age, gender, or place of living.
AD has become nearly twice as prevalent as vascular dementia (VaD) in Korea, Japan, and China since the early 1990s. American and European studies consistently reported AD to be more prevalent than VaD. The dementia prevalence rate was found to be 11.2 per 1,000 among Chinese aged 50 years and older on the islet of Kinmen. AD accounted for 64.6% and VaD for 29.3%. These results, together with previous studies in Chinese populations, suggest that the rates of AD in the Chinese are low compared with those in whites.
In Nigeria, the prevalence of dementia was found to be low. Indian studies have been contradictory, with both AD and VaD being more prevalent in different studies.
Age distribution for Alzheimer disease
The prevalence of AD increases with age. AD is most prevalent in individuals older than 60 years. Some forms of familial early-onset AD can appear as early as the third decade, but familial cases constitute less than 10% of AD overall.
More than 90% of cases of AD are sporadic and occur in individuals older than 60 years. Of interest, however, results of some studies of nonagenarians and centenarians suggest that the risk may decrease in individuals older than 90 years. If so, age is not an unqualified risk factor for the disease, but further study of this matter is needed.
Savva et al found that in the elderly population, the association between dementia and the pathological features of AD (eg, neuritic plaques, diffuse plaques, tangles) is stronger in persons 75 years of age than in persons 95 years of age. These results were achieved by assessing 456 brains donated to the population-based Medical Research Council Cognitive Function and Ageing Study from persons 69-103 years of age at death. [65]
Studies have demonstrated that the relationship between cerebral atrophy and dementia persist into the oldest ages but that the strength of association between pathological features of AD and clinical dementia diminishes. It is important to take age into account when assessing the likely effect of interventions against dementia.
Sexual differences in incidence
Some studies have reported a higher risk of AD in women than in men; other studies, however, including the Aging, Demographics, and Memory Study, found no difference in risk between men and women. [66] Almost two thirds of Americans with AD are women. [10] Among AD patients overall, any sexual disparity may simply reflect women’s higher life expectancy. Among those who are heterozygous for the APOE E4 allele, however, Payami et al found a twofold increased risk in women. [67]
Race-related differences in incidence
AD and other dementias are more common in African Americans than in whites. According to the Alzheimer’s Association, in the population aged 71 years and older, African Americans are almost twice as likely to have AD and other dementias as whites (21.3% of African Americans vs 11.2% of whites). The number of Hispanic patients studied in this age group was too small to determine the prevalence of dementia in this population.
Based on data for Medicare beneficiaries age 65 and older, 6.9% of whites, 9.4% of African Americans, and 11.5% of Hispanics have AD or other dementias, and the prevalence of AD and other dementias is higher in older age groups. [10]
In a study of 1255 people, including both African American (N=173) and Caucasian participants, researchers found that cerebrospinal fluid from African Americans tended to contain lower levels of brain protein tau, a biomarker associated with AD. However, this did not seem to protect them from the disease; they were just as likely as Caucasians to be cognitively impaired in this study. These results suggest that analysis of biomarkers for AD should be adjusted for race. [68]
Prognosis
AD is initially associated with memory impairment that progressively worsens. Over time, patients with AD can also display anxiety, depression, insomnia, agitation, and paranoia. As their disease progresses, patients with AD come to require assistance with basic activities of daily living, including dressing, bathing, and toileting. Eventually, difficulties with walking and swallowing may develop. Feeding may be possible only by gastrointestinal tube, and difficulty swallowing may lead to aspiration pneumonia.
The time from diagnosis to death varies from as little as 3 years to as long as 10 or more years. Patients with early-onset AD tend to have a more aggressive, rapid course than those with late-onset AD. The primary cause of death is intercurrent illness, such as pneumonia.
Patient Education
When counseling patients following a diagnosis of AD, it is essential to involve the patient’s family and others who will play a supporting role in the discussion. It is important to emphasize that not only the patient but also those who support the patients will likely experience reactions of sadness and anger and that these are normal reactions to such a diagnosis.
As the patient’s symptoms become more pronounced, a dialogue must be opened regarding the patient’s wishes for care when he or she is no longer able to make the necessary choices. Durable power of attorney should be discussed, with particular attention to who will make decisions for both medical and financial issues. Medical advance directives should be considered while the patient is still able to participate in the decision-making process.
Throughout the course of the disease, family members should be careful to select qualified and trustworthy individuals to be involved in the day-to-day management of the patient. Caregivers need to balance attention to the patient’s physical needs with maintaining respect for the patient as a competent adult, to the extent allowed by the progression of the disease. Any suspicions of elder abuse should be immediately addressed.
Above all, counseling of AD patients and families should emphasize that patients should continue to engage in activities that they enjoy doing. Maintaining an optimal quality of life is key.
The following resources may be helpful to share with patients and their families:
-
MedlinePlus -Alzheimer’s Disease
-
The National Institute on Aging -General Information on Alzheimer’s Disease
-
Caring for Someone with Alzheimer’s (video series)
For patient education information, see the Dementia Center ,as well as Alzheimer Disease, Alzheimer Disease in Individuals With Down Syndrome, Dementia Overview, and Dementia Medication Overview
-
APP is associated with the cell membrane, the thin barrier that encloses the cell. After it is made, APP sticks through the neuron's membrane, partly inside and partly outside the cell. Image courtesy of NIH.
-
Enzymes (substances that cause or speed up a chemical reaction) act on the APP and cut it into fragments of protein, one of which is called beta-amyloid. Image courtesy of NIH.
-
The beta-amyloid fragments begin coming together into clumps outside the cell, then join other molecules and non-nerve cells to form insoluble plaques. Image courtesy of NIH.
-
Healthy neurons. Image courtesy of NIH.
-
Image courtesy of NIH.
-
Preclinical Alzheimer disease. Image courtesy of NIH.
-
Mild Alzheimer disease. The disease begins to affect the cerebral cortex, memory loss continues, and changes in other cognitive abilities emerge. The clinical diagnosis of AD is usually made during this stage. Image courtesy of NIH.
-
Severe Alzheimer disease. In the last stage of AD, plaques and tangles are widespread throughout the brain, and areas of the brain have atrophied further. Patients cannot recognize family and loved ones or communicate in any way. They are completely dependent on others for care. All sense of self seems to vanish. Image courtesy of NIH.
-
Preclinical Alzheimer disease. Image courtesy of NIH.
-
Mild-to-moderate Alzheimer disease. Image courtesy of NIH.
-
Severe Alzheimer disease. Image courtesy of NIH.
-
Cortical atrophy with hydrocephalus ex vacuo is seen in Alzheimer disease.
-
Plaques and tangles in Alzheimer disease.
-
Amyloid angiopathy in Alzheimer disease.
-
Coronal T1-weighted magnetic resonance imaging (MRI) scan in a patient with moderate Alzheimer disease. Brain image reveals hippocampal atrophy, especially on the right side.