Background
Neuroacanthocytosis (NA) syndromes include combined features of acanthocytosis (ie, spiked red blood cells), chorea, orofacial tics, amyotrophy often with hyperCKemia, and normobetalipoproteinemia. NA has been described as inherited as an autosomal recessive disorder, as an autosomal dominant disorder, and as part of an X-linked disorder called McLeod syndrome (MLS). The autosomal recessive type, usually called chorea-acanthocytosis, is most common and was originally described by Levine and Critchley in the 1960s. [1, 2, 3] In 2001, the gene for this recessive type was characterized on chromosome 9. Since that year, rarer autosomal dominant disease forms with variable penetrance with or without chromosome 9 abnormalities have also been described. In all types, the neurologic course is progressive. Degeneration of the basal ganglia is a consistent feature of this disorder.
All of the syndromes under the NA umbrella are distinguished from the Bassen-Kornzweig syndrome, an autosomal recessive disorder of childhood in which abetalipoproteinemia and acanthocytosis occur along with steatorrhea, retinitis pigmentosa, and cerebellar ataxia.
Acanthocytosis has also been associated with the rare hypobetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP) syndrome, a disease of childhood akin to Hallervorden-Spatz disease and a defect in the gene for pantothenate kinase.
The array of clinical features in NA syndromes is complex. Not only are cases known in which neurologic features of classic adult and childhood acanthocytosis syndromes overlap, but adult forms have been well described in which lipid profiles more closely resemble those of Bassen-Kornzweig syndrome, as have adult forms that begin in childhood.
-
An adult NA syndrome due to an X-linked gene defect is known that largely excludes females.
-
NA syndromes that include parkinsonism, peripheral neuropathy, myopathy, seizures, and psychosis have been described.
-
Adult-type variants of NA have been associated with general medical disorders involving the heart and immune system.
In a detailed pathophysiological study, the well-described choreiform movement disorder of NA has been described coexisting with an associated peripheral neuropathy in a patient without acanthocytosis. See the image below.
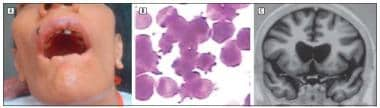 Patient with choreoacanthocytosis. A: Note self-mutilation of the lips owing to orofacial dyskinesia. B: Peripheral blood smear exhibits acanthocytes (Wright-Giemsa, original magnification, >100). C: Coronal view of T1-weighted MRI shows atrophy of the caudate nuclei. Archives of Neurology 64(11):1661-1664 2007 Copyright © 2010 American Medical Association.
Patient with choreoacanthocytosis. A: Note self-mutilation of the lips owing to orofacial dyskinesia. B: Peripheral blood smear exhibits acanthocytes (Wright-Giemsa, original magnification, >100). C: Coronal view of T1-weighted MRI shows atrophy of the caudate nuclei. Archives of Neurology 64(11):1661-1664 2007 Copyright © 2010 American Medical Association.
For related information, see Neuroacanthocytosis.
Pathophysiology
The precise pathophysiology is not understood. Clues to the pathogenesis of the disorder arise from the observation that both the neurological and hematological systems are affected.
In the classic form of the disorder, central nervous system pathologic features include atrophy of the caudate and putamen and, to a lesser extent, the globus pallidus and substantia nigra. A cell loss of 90% in the striatum with astrocytic gliosis has been reported. In contrast to Huntington disease (HD), the major inherited choreiform disorder of adults, the cerebral cortex and corpus callosum are relatively spared. Additionally, the presence of acanthocytosis distinguishes NA from HD.
Defects in such disparate systems (ie, basal ganglia and erythrocytes) have led to the suggestion that a common neurohematological membrane defect is involved.
In 2001, a deletion mutation in the gene (now known as VPS13A) localized to chromosome band 9q21 was identified as the site for the defect generating the autosomal recessive form of NA. It has been determined that VPS13A encodes for a protein called chorein. Thus, patients with NA typically carrying this deletion mutation have a deficiency or even absence of chorein.
In 2005, based upon research involving several large French-Canadian families that presented with temporal lobe epilepsy, an expanded conceptualization of the molecular genetics of the autosomal recessive form NA was attained. Of family members in this research who presented with epilepsy, 70-80% had large deletions in the NA gene, now known as VPS13A, on chromosome 9. Some family members with no epilepsy but with milder features, such as tics and dysphagia for example, may be representative of heterozygous expression of the deletion, suggesting that variations in the VPS13A gene may lead to a dominant pattern of inheritance.
VPS13A may be involved in the control of protein cycling through the trans-Golgi network. It is broadly expressed and found in the brain, heart, skeletal muscle, and kidney.
The chorein deficiency has been also linked to upregulation of gephyrin and GABA(A) receptors.
Japanese researchers support this latter point by positing that the disorder in several families studied with the chorein defect and no seizures may have a dominant form of NA with incomplete penetrance. Further genetic variability is derived from the work of Walker who found an autosomal dominant NA family with a Huntington disease–like syndrome (HDL2) characterized by a defect in the junctophilin-3 gene and not the chorein gene. [4]
To further induce pathophysiological consternation, the McLeod syndrome (MLS) seen overwhelmingly in males has many features akin to the autosomal forms and is due to a completely separate abnormality featuring the following: (1) absent expression of Kx erythrocyte antigen, (2) weak expression of Kell glycoprotein antigens, (3) universally present hyperCKemia, and (4) X-linked inheritance. (Recently, the Kx protein has been shown to be neuronal, located mainly in intracellular compartments, suggesting a cell specific trafficking pattern.)
Variation in other systems in patients with NA syndromes reflects the possibility of genetic heterogeneity that is more wide ranging than what may be noted in the affected components of the red blood cell (RBCs) and striatum alone.
Indeed, Walker et al [5] noted in 2011 that in NA there exist several associated genetic loci in the 9q21 region. Many defects, including gene deletions and insertions, as well as missense, nonsense, and splicing, mutations, have been found spread over hundreds of kilobases of genomic DNA, making the authors' case for exome sequencing to help with the diagnosis and genetic counseling, including carrier detection.
Other common sites of pathophysiological dysfunction are the spinal cord, muscles, and nerves.
Evidence of denervation with fasciculations has been noted intermittently on electromyography (EMG) and is consistent with motor neuron disease despite absence of anterior horn cell histopathology.
Neurogenic muscle atrophy on muscle biopsy is consistent with a possible insult affecting the anterior horn cells of the spinal cord or their axons, although a primary myopathy and even myositis also have been described.
The consistently noted increase in creatine phosphokinase (CPK) level may be due to a primary myopathy, neurogenic atrophy, or chorea. Chorein deficiency in NA has been implicated in the cause of the myopathy.
Nerve biopsy has revealed loss of large myelinated axons consistent with a distal axonopathy.
Both RBC membrane protein and lipid abnormalities have been described, notably in the critical band 3 protein layer (most recently in the Walker family) and in an abnormal composition of covalently bound fatty acids.
Antibodies to the GM1 ganglioside component of peripheral nerves have been described. This GM1 ganglioside is also present in RBC membranes and in the central nervous system. Decreases in GM3 and sialoparagloboside components of RBC membranes have been noted. These gangliosides are also present throughout the nervous system.
Many of the patients with MLS have cardiomyopathy or hemolytic anemia, features not as commonly noted in the autosomal cases.
Redman and Reid have commented on the complexity of the Kell blood group proteins whereby the Kell protein expressed via a gene on chromosome 7 interacts with the XK protein, strikingly absent in patients with MLS. [6] These proteins are preferentially expressed in erythroid tissue but are also present in lesser amounts in brain and skeletal and cardiac muscle. The Kell protein is essential in the activation of the endothelin system and is important in cell membrane integrity. The XK protein bound to it in a 2-protein complex may have a complementary role as a membrane transporter. Experimental evidence cited by van den Buuse and Webber suggests endothelins may be basal ganglia neurotransmitters. [7] Thus, the implication exists for a neurochemical tie to the NA syndromes, so often highlighted by basal ganglia dysfunction.
Jung et al have noted that in MLS, known disease-causing XK gene mutations comprised deletions, nonsense, or splice-site mutations, predicting absent or truncated XK protein devoid of the Kell-protein–binding site, but they found 2 brothers who had XK missense mutation without hematologic, neuromuscular, or cerebral involvement. However, despite their having the McLeod phenotype, the mutated XK protein seemed to be largely functional. [8]
Meanwhile, speaking to the great variability in MLS disorders, Wiethoff, in 2013 described a 70-year-old man of Greek origin with choreatic movements of the tongue and face, lower limb muscle weakness, peripheral neuropathy, elevated creatine phosphokinase (CPK), acanthocytosis, and hemolysis in the absence of Kell RBC antigens with an additional factor IX deficiency. Genetic testing for mutations in the 3 exons of the XK gene revealed a previously unreported hemizygous single base-pair frameshift deletion at exon 1 (c.229delC, p.Leu80fs). [9]
Bosman and De Franceschi have also summarized several studies of non-McLeod NA that have shown abnormalities in the aforementioned band 3 region. [10, 11] Changes in band 3 structure do not only lead to alterations in erythrocyte shape but also to altered anion transport characteristics and increased age-related autoimmunoreactivity, with anti-band 3 antibodies noted in patients with NA. Elaborating on this latter point, echinocytes are normally aging misshapen RBCs reported to have band 3 abnormalities as well.
Brain band 3 change is also tied to neuronal degeneration and has been linked generally to extrapyramidal movement disorders and axonal neuropathies.
These insights, though incompletely understood, suggest that the pathophysiology of all of the NA syndromes involves different gene abnormalities that can cause multisystem membrane defects. The common derangement is in the malformation of the RBC shape and the induction of various levels of central nervous system, neuromuscular, and cardiac dysfunction. Intriguingly is the prospect that some kind of accelerated senescence and autoimmune damage to both erythrocytes and nerve tissue holds a key in fully appreciating the triggering of acanthocytosis and neurodegeneration in NA syndromes.
Mindful that the neuroacanthocytotic MLS and a recently described non-McLeod NA family with no typical autosomal recessive gene NA defect are not due to a specific chorein protein abnormalities, it is still extremely important to expand our knowledge of chorein, the protein specifically linked to most cases of NA. Chorein is normally present in man. Dobson-Stone suggests the CHAC or chorein gene locus is abnormal in many ways to induce NA by either not producing gene product or yielding a truncated nonfunctional protein. [12]
However, beyond being involved in protein-protein trafficking, how this protein leads to malconfigured erythrocytes and the array of neuropathological and clinical signs of NA is not clear. A patient has been described with chorein deficiency in red blood cells in the absence of acanthocytosis. [13] Further confounding the issue, deficiency of erythroid 4.1R protein has been described in patients with NA. This protein is distinct from chorein and essential for maintaining erythrocyte shape and mechanical properties of the membrane, such as deformability and stability.
Many issues in NA and MLS are still unresolved, not the least of which is why these 2 disorders present syndromes that are so similar, despite showing distinct genetic defects. Why the genetic defects in NA and MLS induce hematologic, cardiac, and neurologic abnormalities is also not clear. In MLS, Walker and Danek note that different Kell mutations may have different effects on the Kell gene product and thus may account for the variable phenotype in patients with MLS. Indeed, this variable mutation phenomenon may explain the differing clinical presentations in the autosomal gene NA syndromes (non-MLS). [14]
In 2011, De Franceschi et al [15] published a report that indicated erythrocyte membrane changes of NA patients are the result of altered Lyn kinase (LYN) activity, which is involved in modulating band 3 function on the RBC membrane. [16]
In 2012, De Franceschi et al [17] went beyond the LYN identification and attributed NA acanthocyte genesis to a very restricted group of highly interconnected kinases, including LYN and ABL1, ABL2, AURKA, CDK5, EPHB2, EPHB4, FYN, MAP4K2, MAPK14, PDPK1, RPS6KA3, TGFBR1, and TTN, that regulate rho small GTPase-mediated signaling, cytoskeleton network, erythropoiesis, and neurogenesis. The authors maintain that this network may represent a shared regulatory cluster of kinases whose alteration is most likely involved in the generation of the abnormal red blood cells that characterize NA. They also believe these same kinases might be responsible for acanthocyte generation in MLS.
Epidemiology
Frequency
United States
NA syndromes have been described in consanguineous and nonconsanguineous families of English and Puerto Rican descent.
International
NA syndromes have been described in American (USA), Chinese, Japanese, Malaysian, South-African black, Mexican, Brazilian, British, Spanish, Portuguese, Australian, Indian, Italian, Chilean, German, Turkish, Scandinavian, French-Canadian, French, and Thai [18] populations.
MLS has been described in multiple nationalities and races, with a Chinese patient first described with it in 2013 [19] and the first patients (a family) from India first described with the disorder in 2011. [20]
Mortality/Morbidity
NA syndromes are often fatal.
A common cause of death is aspiration pneumonia due to movement disorder-induced impairment in swallowing.
Other causes of death include complications of cardiomyopathy and suicide as a result of depression or psychosis.
Morbidity is related specifically to the progressive movement disorder and muscle wasting.
Malnutrition is very common in many of these neurological syndromes.
Although the acanthocytosis often is noted spectacularly on peripheral blood smear (approaching 50% of patients) it usually is not associated with hemolytic anemia or other life-threatening hematological problems. However, hemolysis has been described, which can carry significant morbidity.
Race
NA syndromes have been described in all races.
Sex
Overall, NA syndromes are more common in men (partly due to the McLeod syndrome types, which are X-linked and therefore almost exclusively found in men).
Presumed autosomal recessive NA is more common in males, with a male-to-female ratio as high as 70:30.
Age
The adult-type NA syndromes usually begin in mid life (age 20-50 y). However, they also have been reported to occur in childhood.
-
Patient with choreoacanthocytosis. A: Note self-mutilation of the lips owing to orofacial dyskinesia. B: Peripheral blood smear exhibits acanthocytes (Wright-Giemsa, original magnification, >100). C: Coronal view of T1-weighted MRI shows atrophy of the caudate nuclei. Archives of Neurology 64(11):1661-1664 2007 Copyright © 2010 American Medical Association.








