Practice Essentials
Pantothenate kinase-associated neurodegeneration (PKAN), formerly called Hallervorden-Spatz Disease (HSD), is a rare disorder characterized by progressive extrapyramidal dysfunction and dementia. The disease was first described in 1922 as a form of familial brain degeneration characterized by cerebral iron deposition.
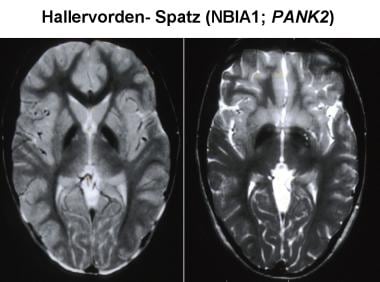 Magnetic resonance imaging (MRI) has increased the likelihood of antemortem diagnosis of Pantothenate kinase-associated neurodegeneration (PKAN). The typical MRI findings include bilaterally symmetrical, hyperintense signal changes in the anterior medial globus pallidus, with surrounding hypointensity in the globus pallidus, on T2-weighted images. These imaging features, which are fairly diagnostic of PKAN, have been termed the "eye-of-the-tiger sign." The hyperintensity represents pathologic changes, including gliosis, demyelination, neuronal loss, and axonal swelling. The surrounding hypointensity is due to loss of signal secondary to iron deposition.
Magnetic resonance imaging (MRI) has increased the likelihood of antemortem diagnosis of Pantothenate kinase-associated neurodegeneration (PKAN). The typical MRI findings include bilaterally symmetrical, hyperintense signal changes in the anterior medial globus pallidus, with surrounding hypointensity in the globus pallidus, on T2-weighted images. These imaging features, which are fairly diagnostic of PKAN, have been termed the "eye-of-the-tiger sign." The hyperintensity represents pathologic changes, including gliosis, demyelination, neuronal loss, and axonal swelling. The surrounding hypointensity is due to loss of signal secondary to iron deposition.
Signs and symptoms
Symptoms in PKAN include the following:
-
Dystonia - A prominent and early feature
-
Significant speech disturbances - Can occur early
-
Dysphagia - A common symptom; caused by rigidity and corticobulbar involvement
-
Dementia - Present in most individuals with PKAN
-
Visual impairment - Caused by optic atrophy or retinal degeneration; not uncommon and can be the presenting symptom of the disease, although this is rare
-
Seizures - Have been described [18]
Clinical manifestations of PKAN vary from patient to patient. The symptoms usually begin in the first decade with a motor disorder of extrapyramidal type and gait difficulty. Extrapyramidal symptoms dominate the clinical picture and include rigidity, slowness of movement, dystonia, choreoathetosis, and tremor.
Diagnosis
No biochemical markers have been found in PKAN. Levels of copper, ceruloplasmin, lipids, amino acids, and acanthocytes typically are measured in the blood to exclude other conditions. Radionuclide scan reveals increased iron uptake in the basal ganglia. [23]
Management
Historically, the treatment of patients with PKAN was mostly symptomatic. However, novel therapies have been studied as potential disease-modifying agents. These include iron chelators as well as fosmetpantotenate.
In terms of symptomatic therapy, tremor responds best to dopaminergic agents. The anticholinergic agent benztropine may be used to help rigidity and tremor. Benzodiazepines have been tried for choreoathetotic movements.
Background
Pantothenate kinase-associated neurodegeneration (PKAN), formerly called Hallervorden-Spatz Disease (HSD), is a rare disorder characterized by progressive extrapyramidal dysfunction and dementia. The disease was first described in 1922 by two German physicians, Hallervorden and Spatz, as a form of familial brain degeneration characterized by cerebral iron deposition. The term neurodegeneration with brain iron accumulation type 1 (NBIA-1), eventually came to be used for this condition, [1, 2] although the most recent term for the disorder is pantothenate kinase-associated neurodegeneration (see the image below). (See Etiology.) [3]
Magnetic resonance imaging (MRI) has increased the likelihood of antemortem diagnosis of Pantothenate kinase-associated neurodegeneration (PKAN). The typical MRI findings include bilaterally symmetrical, hyperintense signal changes in the anterior medial globus pallidus, with surrounding hypointensity in the globus pallidus, on T2-weighted images. These imaging features, which are fairly diagnostic of PKAN, have been termed the "eye-of-the-tiger sign." The hyperintensity represents pathologic changes, including gliosis, demyelination, neuronal loss, and axonal swelling. The surrounding hypointensity is due to loss of signal secondary to iron deposition.
The onset of symptoms most commonly occurs in late childhood or early adolescence. The classic presentation is in the late part of the first decade or the early part of the second decade, between ages 7 and 15 years. However, the disease has been reported in infancy, and cases with adult onset have also been described. (See Presentation and Workup.) [4, 5, 6]
PKAN is relentlessly progressive. The clinical course is characterized by progressive dementia, spasticity, rigidity, dystonia, and choreoathetosis. The progression of the disease usually occurs over 10-12 years, and affected individuals typically die in the second or third decade; however, case reports describe patients surviving 30 years. (See Presentation and Prognosis.) [7, 8]
The disease can be familial or sporadic. When familial, PKAN is inherited recessively; it has been linked to chromosome 20. [9] A mutation in the pantothenate kinase (PANK2) gene on band 20p13 has been described in patients with PKAN. (See Etiology.) [10]
PKAN is a subtype of Neurodegeneration with Brain Iron Accumulation (NBIA). NBIA comprises a group of inherited neurodegenerative disorders that are characterized symptomatically by extrapyramidal movement disorders and pathologically by abnormal iron accumulation in deep basal ganglia. Ten forms have been described so far, and each has a corresponding gene and specific mode of inheritance. PKAN, PLA2G6-Associated Neurodegeneration (PLAN), Fatty Acid Hydroxylase-associated Neurodegeneration (FAHN), Coenzyme A synthase Protein-Associated Neurodegeneration (CoPAN), Mitochondrial-membrane Protein-Associated Neurodegeneration (MPAN), Kufor-Rakeb syndrome, Woodhouse-Sakati syndrome, and Aceruloplasminemia are all autosomal recessive; whereas Beta-propeller Protein-Associated Neurodegeneration (BPAN) is X-linked dominant and Neuroferritinopathy is autosomal dominant.
In the United States, PKAN is the most common form of NBIA, comprising 50% of all NBIA cases. PLAN is the second most common, comprising 20% of all cases. [11]
Etiology
The exact etiology of PKAN is not known. One proposed hypothesis is that abnormal peroxidation of lipofuscin to neuromelanin and deficient cysteine dioxygenase lead to abnormal iron accumulation in the brain. While portions of the globus pallidus and pars reticulata of substantia nigra (SN) have high iron content in healthy individuals, individuals with PKAN have excess amounts of iron deposited in these areas. [3]
However, the exact role of iron in the etiology of this disease remains unknown. Also, whether the deposition of iron in basal ganglia in PKAN is the cause or consequence of neuronal loss and gliosis is not clear. Decreased activity of the enzyme cysteine dioxygenase was demonstrated in one affected child. [12] This was postulated to lead to accumulation of cysteine in the basal ganglia, since cysteine can chelate iron and thus result in its deposition. However, these findings were not confirmed in adult patients.
A role for mutation in the PANK2 gene (band 20p13) in the etiology of PKAN has been proposed. Deficiency of pantothenate kinase may lead to accumulation of cysteine and cysteine-containing compounds in the basal ganglia. This causes chelation of iron in the globus pallidus and free radical generation as a result of rapid auto-oxidation of cysteine in the presence of iron. [13] Mutations in the PANK2 gene account for most inherited PKAN cases. Such mutations result in an autosomal recessive inborn error of coenzyme A metabolism, which has been termed pantothenate kinase–associated neurodegeneration. [14, 15, 16, 17, 18]
Pathologic examination reveals characteristic rust-brown discoloration of the globus pallidus and SN pars reticulata secondary to iron deposition. [19, 20] Generalized atrophy of the brain may be noted, with a reduction in size of the caudate nuclei, SN, and tegmentum. Microscopically, the characteristic changes include the following:
-
Variable loss of neurons, myelinated fibers, and gliosis in globus pallidus and SN, which may appear spongiotic when severe
-
Widely disseminated rounded or oval, nonnucleated structures called spheroids (also known as axon schollen or neuroaxonal dystrophy); these represent swollen axons with vacuolated cytoplasm and are found most abundantly in the pallidonigral system, although they are also present in the cerebral cortex
-
Accumulation of pigment, mostly containing iron, in the globus pallidus and SN
-
Ceroid lipofuscin and neuromelanin containing iron, in the areas mainly affected (mentioned above)
Iron deposition may be found intracellularly and extracellularly and frequently is centered on vessels. These changes are found to a lesser degree in other parts of the brain and in the spinal cord. [21] The presence of axonal spheroids suggests a link between PKAN and infantile neuroaxonal dystrophy; however, no clinical or genetic relationship has been reported between the 2 diseases. Tau-positive neurofibrillary tangles and alpha-synuclein–positive Lewy bodies may be found in cortical and subcortical regions in patients with a prolonged clinical course. [1]
Prognosis
The clinical course of PKAN is variable, depending on its form (classic or atypical). In patients with the classic form, the disease has a progressive course extending over several years, leading to death in early childhood. Some patients experience rapid deterioration of function secondary to dystonia, rigidity, dysphagia, and respiratory compromise and die within 1–2 years of disease onset. Other patients undergo a slower progression or even plateau for many years and may continue to function into the third decade of life. [8]
-
Magnetic resonance imaging (MRI) has increased the likelihood of antemortem diagnosis of Pantothenate kinase-associated neurodegeneration (PKAN). The typical MRI findings include bilaterally symmetrical, hyperintense signal changes in the anterior medial globus pallidus, with surrounding hypointensity in the globus pallidus, on T2-weighted images. These imaging features, which are fairly diagnostic of PKAN, have been termed the "eye-of-the-tiger sign." The hyperintensity represents pathologic changes, including gliosis, demyelination, neuronal loss, and axonal swelling. The surrounding hypointensity is due to loss of signal secondary to iron deposition.








