Practice Essentials
The term chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) has been used to identify patients with a chronically progressive or relapsing symmetric sensorimotor disorder with cytoalbuminologic dissociation and interstitial and perivascular endoneurial infiltration by lymphocytes and macrophages. It can be considered the chronic equivalent of acute inflammatory demyelinating polyradiculoneuropathy, the most common form of Guillain-Barré syndrome.
Signs and symptoms
CIDP typically starts insidiously and evolves slowly, in either a slowly progressive or a relapsing manner, with partial or complete recovery between recurrences; periods of worsening and improvement usually last weeks or months. Most experts consider the necessary duration of symptoms to be greater than 8 weeks for the diagnosis of CIDP to be made.
Symptoms reported include the following:
-
Preceding infection (infrequent)
-
Initial limb weakness, both proximal and distal
-
Sensory symptoms (eg, tingling and numbness of hands and feet)
-
Motor symptoms (usually predominant)
-
In about 16% of patients, a relatively acute or subacute onset of symptoms
-
In children, usually a more precipitous onset of symptoms
-
Symptoms of autonomic system dysfunction (eg, orthostatic dizziness)
Pertinent physical findings are limited to the nervous system, except when the condition is associated with other diseases. Such findings may include the following.
-
Signs of cranial nerve (CN) involvement (eg, facial muscle paralysis or diplopia)
-
Gait abnormalities
-
Motor deficits (eg, symmetric weakness of both proximal and distal muscles in upper and lower extremities)
-
Diminished or absent deep tendon reflexes
-
Sensory deficits (typically in stocking-glove distribution)
-
Impaired coordination
See Clinical Presentation for more detail.
Diagnosis
Laboratory studies that may be helpful include the following:
-
Cerebrospinal fluid analysis: Elevated protein levels are common (80% of patients); 10% of patients also have mild lymphocytic pleocytosis and increased gamma globulin
-
Complete blood count (CBC), erythrocyte sedimentation rate (ESR), antinuclear antibody (ANA) level, biochemistry profile, and serum and urine immunoelectrophoresis (to exclude associated systemic disorders)
-
In certain instances, genetic testing
Other tests and procedures that may be warranted are as follows:
-
MRI of the spine with gadolinium enhancement
-
Electromyography (EMG) is a critical test to determine whether the disorder is truly a peripheral neuropathy and whether the neuropathy is demyelinating
-
Peripheral (sural) nerve biopsy (see the image below): This is considered when the diagnosis is not completely clear, when other causes cannot be excluded, or when profound axonal involvement is observed on EMG; biopsy was once commonly recommended for most patients before immunosuppressive therapy, but current guidelines no longer recommend it
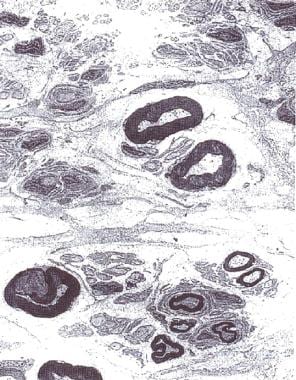 Electron micrograph of the peripheral nerve of a patient with chronic inflammatory demyelinating polyradiculoneuropathy. Note "onion bulb" formation in the myelin sheath of the nerve fibers due to continuous demyelination and remyelination. Courtesy of A. Sima, MD, Department of Pathology, Wayne State University.
Electron micrograph of the peripheral nerve of a patient with chronic inflammatory demyelinating polyradiculoneuropathy. Note "onion bulb" formation in the myelin sheath of the nerve fibers due to continuous demyelination and remyelination. Courtesy of A. Sima, MD, Department of Pathology, Wayne State University.

See Workup for more detail.
Management
Principles of treatment are as follows:
-
CIDP must be treated to prevent accumulating disability that necessitates physical and occupational therapy, orthotic devices, and long-term treatment
-
Close follow-up care is necessary to adjust treatment
-
Surgical and orthopedic consultation may be required for sural nerve biopsy or in severe disease with joint deformities
-
Consultation with a neurologist is warranted
-
Consultation with a physical medicine and rehabilitation specialist is appropriate for physical and occupational therapy and evaluation for orthotic devices
-
Other consultations may be necessary if associated diseases are present
-
Physical therapy and an active lifestyle should be encouraged
See Treatment and Medication for more detail.
Background
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is presumed to occur because of immunologic antibody-mediated reaction along with interstitial and perivascular infiltration of the endoneurium with inflammatory T cells and macrophages. The consequence is a segmental demyelination of peripheral nerves.
Human leukocyte antigens Dw3, DRw3, A1, and B8 occur more frequently in patients with CIDP than in the healthy population.
Cytoalbuminologic dissociation is a characteristic finding in cerebrospinal fluid (CSF) pointing to nerve root involvement. Occasionally, CSF studies reveal mild lymphocytic pleocytosis and elevation of gamma globulin level, but this is observed most frequently in HIV-positive patients.
The term chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) has been used to identify patients with a chronically progressive or relapsing symmetric sensorimotor disorder with cytoalbuminologic dissociation and interstitial and perivascular endoneurial infiltration by lymphocytes and macrophages. In many ways, CIDP can be considered the chronic equivalent of acute inflammatory demyelinating polyradiculoneuropathy (AIDP), the most common form of Guillain-Barré syndrome (GBS).
A number of variants of CIDP have been described that have immune or inflammatory aspects and electrophysiologic and/or pathologic evidence of demyelination in common. No consensus exists on the best approach to the nomenclature of these disorders. CIDP is a major subset of chronic acquired demyelinating polyneuropathies (CADP). In this context, CIDP is considered when patients have a symmetric proximal and distal motor predominant disorder.
CIDP variants include patients with predominantly sensory symptoms, those with a distal symmetric disorder (DADS), those with multifocal sensorimotor neuropathy or sensorimotor mononeuropathy multiplex with prominent conduction block (also known as Lewis-Sumner neuropathy), and those with CIDP with associated CNS demyelination or with other systemic disorders.
The following disorders are considered distinct from CIDP because they have specific pathophysiologic features and respond to treatments differently than do patients with CIDP: Demyelinating neuropathies associated with immunoglobulin M (IgM) paraproteins, including those with anti–myelin-associated glycoprotein (MAG) antibodies; polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes (POEMS) syndrome; and multifocal motor neuropathy.
Pathophysiology
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is presumed to occur because of immunologic antibody-mediated reaction along with interstitial and perivascular infiltration of the endoneurium with inflammatory T cells and macrophages. The consequence is a segmental demyelination of peripheral nerves.
Human leukocyte antigens Dw3, DRw3, A1, and B8 occur more frequently in patients with CIDP than in the healthy population.
Cytoalbuminologic dissociation is a characteristic finding in cerebrospinal fluid (CSF) pointing to nerve root involvement. Occasionally, CSF studies reveal mild lymphocytic pleocytosis and elevation of gamma globulin level, but this is observed most frequently in HIV-positive patients.
Epidemiology
Frequency
CIDP is uncommon. The estimated prevalence of CIDP in populations from the UK, Australia, Italy, Norway, and Japan is 0.8-7.7 per 100,000. A 2009 study showed that the incidence and prevalence is variable depending on diagnostic criteria. In Rutland, UK on May 1, 2008, the prevalence of CIDP was 4.77/100,000 if the EFNS/PNS criteria were used but only 1.97 per 100,000 if the AAN criteria were used. Similarly the annual incidence was 0.7 per 100,000 using the EFNS criteria and 0.35 using the AAN criteria. [1]
Mortality/morbidity
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) most commonly has an insidious onset and either chronic progressive or relapsing course. Occasionally, complete remissions occur. Quadriplegia, respiratory failure, and death have been described but are rare.
Race-, sex-, and age-related demographics
No racial predilection has been identified.
Both sexes are affected. Of CADP variants, multifocal motor neuropathy has a male predominance of at least 2:1 based on a survey of the largest case series.
Chronic inflammatory demyelinating polyradiculoneuropathy may occur at any age, but it is more common in the fifth and sixth decades. Relapsing course is associated with younger age of patients (third and fourth decades). CIDP has been described in childhood.
Prognosis
Some have suggested that patients with relapsing chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) have a better prognosis than patients with the chronic progressive course. Approximately 70% of patients are said to make relatively good recovery from their relapses, and close to 90% of patients respond to initial immunosuppressive therapy. Some patients do not respond to the usual treatments and accumulate significant disability. Some patients have only a short treatment effect and become treatment dependent. A useful way of understanding the clinical status of patients is to use the CIDP activity status (CDAS). This approach has been useful for both clinical research and for clinical practice. [2]
-
Electromyography of a patient with chronic inflammatory demyelinating polyradiculoneuropathy illustrating conduction block, temporal dispersion of compound muscle action potential, prolonged distal latencies, and slowed conduction.
-
Prolonged F wave latencies (normal is < 31).
-
Electron micrograph of the peripheral nerve of a patient with chronic inflammatory demyelinating polyradiculoneuropathy. Note "onion bulb" formation in the myelin sheath of the nerve fibers due to continuous demyelination and remyelination. Courtesy of A. Sima, MD, Department of Pathology, Wayne State University.






