Practice Essentials
Normal immunoglobulin molecules are symmetrical and consist of 2 pairs of polypeptide chains, designated the light and heavy chains, which are interconnected by disulfide bonds (see the image below). The heavy chains comprise the larger polypeptide subunits, and are specific and distinctive structures that distinguish the major classes of immunoglobulins.
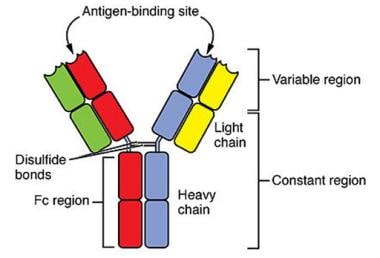 Normal immunoglobulin molecules consist of 2 pairs of polypeptide chains, designated the light and heavy chains, which are interconnected by disulfide bonds. Courtesy of OpenStax (Rice University).
Normal immunoglobulin molecules consist of 2 pairs of polypeptide chains, designated the light and heavy chains, which are interconnected by disulfide bonds. Courtesy of OpenStax (Rice University).
Heavy chain diseases (HCDs) are rare variants of B-cell lymphomas that characteristically involve production of a mutated immunoglobulin heavy chain that is incapable of either partnering with light chains to form a complete immunoglobulin molecule or being eliminated via proteasomal degradation. Their defining feature is the presence of an abnormal heavy chain—immunoglobulin alpha (IgA), gamma (IgG), or mu (IgM)—in the serum without an associated light chain. In addition, free light chain is sometimes present in urine. [1]
The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours classifies the HCDs with plasma cell neoplasms and other diseases with paraproteins under the broader classification of B-cell lymphoid proliferations and lymphomas. [2] The frequency of alpha-, gamma-, and mu-HCD ranges from rare to very rare, respectively. [3] No case of ε-HCD (IgE) or δ-HCD (IgD) has been reported to date.
Ballard and colleagues first described mu-HCD in 1970. [4] Clinical manifestations range from an asymptomatic presentation to aggressive lymphoma (see Presentation). [5] Diagnosis is confirmed by immunofixation that is positive for anti-mu but negative for anti-kappa or anti-lambda light chains (see Workup). [6]
Because of the paucity of cases, no standard treatment has been established for mu-HCD. Currently, the identification of a mu-HCD protein in the serum of an apparently healthy patient should be considered a monoclonal gammopathy of undetermined significance, and the patient should be monitored closely for the development of a symptomatic lymphoproliferative disorder. Once a lymphoproliferative disorder develops, chemotherapeutic agents are used as appropriate for the patient's disorder, stage, and clinical situatio. See Treatment.
This article focuses on mu-HCD; other heavy chain diseases are described elsewhere (see Gamma Heavy Chain Disease).
Pathophysiology
The pathogenesis of mu-HCD is not completely understood. No viral, chemical, physical, or genetic factors have been identified. [7] Most patients have a concurrent lymphoproliferative disorder resembling chronic lymphocytic leukemia/small lymphocytic leukemia. There are also single reports of mu-HCD associated with myelodysplastic syndrome, amyloidosis, and diffuse large B-cell lymphoma.
In mu-HCD the altered heavy chains contain deletions, insertions, and point mutations that are acquired during the process of somatic hypermutation. These alterations typically result in loss of a large portion of the constant-1 (CH1) domain of the heavy chain, which is responsible for light chain binding. Additionally, this abnormal CH1 domain is unable to bind to the heat-shock protein 78 (hsp78), which would otherwise promote proteosomal degradation of the aberrant, unbound heavy chain. The heavy chain thus bypasses elimination and is secreted into the serum, where it can be detected. [1, 8, 9]
Case reports have identified MYD88 mutations in patients with mu-HCD, demonstrating a possible relationship with lymphoplasmacytic lymphoma (LPL). a disorder that commonly presents as Waldenström macroglobulinemia (WM). [10]
Epidemiology
Mu-HCD is the least common of the HCDs. Fewer than 50 cases have been reported in the literature. [11] However, many cases likely have not been reported, especially in the past decade. Given the difficulty in diagnosing this disorder, most reports are from the United States, Western Europe, and Scandinavia.
Mu-HCD is predominantly reported in white men in the fifth or sixth decade of life. However, reports also exist in Black and Asian patients, with ages of diagnosis ranging from 15-80 years.
Prognosis
The course of mu-HCD is variable. Survival ranges from a few months to several years, with a median survival of 24 months in well-studied cases. [6] Possible complications include pathologic fracture secondary to osteolytic lesions. Kidney insufficiency occasionally is associated with mu-HCD, secondary to such disorders as cast glomerulopathy, nodular glomerulosclerosis, and amyloidosis. Cytopenias requiring transfusion are possible in advanced disease.
-
Normal immunoglobulin molecules consist of 2 pairs of polypeptide chains, designated the light and heavy chains, which are interconnected by disulfide bonds. Courtesy of OpenStax (Rice University).




