Practice Essentials
Hairy cell leukemia (HCL) is a chronic lymphoid leukemia, originally described in 1958 by Bouroncle and colleagues [1, 2] and named after the hairlike cytoplasmic projections seen on the surface of the abnormal B-cells (see the image below).
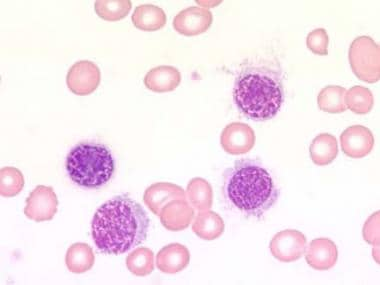 Blood film at × 1000 magnification. This image demonstrates lymphocytes with characteristic cytoplasmic projections. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Blood film at × 1000 magnification. This image demonstrates lymphocytes with characteristic cytoplasmic projections. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
See Chronic Leukemias: 4 Cancers to Differentiate, a Critical Images slideshow, to help detect chronic leukemias and determine the specific type present.
Hairy cell leukemia is recognized as a clonal B-cell malignancy, as identified by immunoglobulin gene rearrangements that result in a phenotype B-cell expression of surface antigens. These reflect the differentiation between the immature B-cell of chronic lymphocytic leukemia and the plasma cell of multiple myeloma.
For patient education information, see the Cancer Center, as well as Leukemia.
Pathophysiology
The abnormal cell in hairy cell leukemia (HCL) is a clonal B-cell lymphocyte (see image below). This cell infiltrates the reticuloendothelial system and interferes with bone marrow function, resulting in bone marrow failure or pancytopenia. [3, 4] The hairy cell also infiltrates the liver and spleen, resulting in organomegaly.
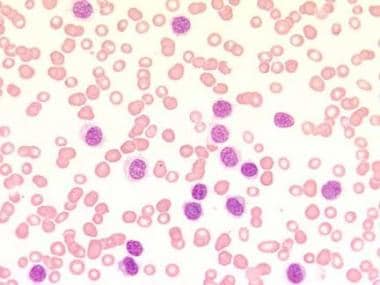 Blood film at × 400 magnification. This image demonstrates a lymphocytosis and an absence of any other type of blood cell (pancytopenia). The characteristic cytoplasmic projections are already visible. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Blood film at × 400 magnification. This image demonstrates a lymphocytosis and an absence of any other type of blood cell (pancytopenia). The characteristic cytoplasmic projections are already visible. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
The pathogenesis of HCL was clarified by the discovery of its underlying genetic cause, the BRAF V600E kinase-activating mutation, which is somatically and clonally present in almost all patients through the entire disease spectrum and clinical course. [5] By aberrantly activating the RAF-MEK-ERK signaling pathway, BRAF V600E shapes key biologic features of HCL, including its specific expression signature, hairy morphology, and antiapoptotic behavior. Accompanying mutations of the KLF2 transcription factor or the CDKN1B/p27 cell cycle inhibitor are recurrent in 16% of patients with HCL and likely cooperate with BRAF V600E in HCL pathogenesis. [6]
The etiology of HCL has not been determined, although some investigators suggest that exposures to benzene, organophosphorus insecticides, or other solvents may be related to disease development. This hypothesis has not been confirmed by other reports, although a French study that evaluated occupational exposure to pesticides and lymphoid neoplasms in men appears to support the hypothesis that occupational pesticide exposures may not only be involved in HCL, Hodgkin lymphoma, and multiple myeloma, but also may play a role in non-Hodgkin lymphoma. [7] Further research is needed.
Other suggested etiologic associations include exposure to radiation, agricultural chemicals, and wood dust, and a previous history of infectious mononucleosis.
Overexpression of cyclin D1 protein, an important cell-cycle regulator, has been observed in HCL and may play a role in the molecular pathogenesis of the disease.
Accumulation of hairy cells in the bone marrow, liver, and spleen, with very little lymph node involvement, is characteristic of HCL. This pattern probably results from the expression of the integrin receptor alpha4-beta1 by the hairy cells and the interaction of the receptor with the vascular adhesion molecule-1 (VCAM-1) found in splenic and hepatic endothelia, bone marrow, and splenic stroma.
Epidemiology
Hairy cell leukemia is relatively uncommon, accounting for 2% of all leukemia cases. An estimated 1100 new cases were diagnosed in the United States in 2016. [8] Hairy cell leukemia is observed more commonly in whites. It has been reported to occur in Ashkenazi Jews, and it is rare in Japan and in people of African descent.
The disease occurs predominantly in middle-aged men, with a median age of 49-51 years. The male-to-female ratio is 4:1 to 5:1. [2, 9]
Prognosis
Hairy cell leukemia behaves like a chronic leukemia. With treatment, most patients achieve clinical remissions and, sometimes, long-term cures. In the United States, 5-year survival ranged from 84% to 94% (lower in Blacks than in Whites). [8]
The risk of second malignancies has been observed in patients with hairy cell leukemia, either through the disease itself or secondary to the immunosuppressive effects of the therapy. Skin cancers (melanoma and non-melanoma) are the most common, representing 33-36% of all secondary malignancies. Other malignancies include prostate cancers, gastrointestinal malignancies, non-Hodgkin lymphomas, and ovarian, cervical, and breast cancer. [10]
A 20-year follow-up in 117 patients in British Columbia showed that 31% developed a second malignancy, of which 30% were diagnosed before hairy cell leukemia was found. [11] On the other hand, an MD Anderson study reported no excess of second malignancies among 350 patients with hairy cell leukemia who were treated with interferon alfa, cladribine, or pentostatin. [12]
-
Blood film at × 400 magnification. This image demonstrates a lymphocytosis and an absence of any other type of blood cell (pancytopenia). The characteristic cytoplasmic projections are already visible. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Blood film at × 1000 magnification. This image demonstrates lymphocytes with characteristic cytoplasmic projections. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Blood film at × 1000 magnification. This image demonstrates tartrate-resistant acid phosphatase (TRAP) activity of lymphocytes. Photographed by U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.







