Practice Essentials
Glycogen storage disease (GSD) VII (Tarui disease) is an autosomal recessive disorder caused by a deficiency of phosphofructokinase (PFK), the enzyme that catalyzes the rate-limiting step in glycolysis. [1] The diagnosis is made on the basis of findings from the history and physical examination, muscle biopsy, electromyography, ischemic forearm testing, and creatine kinase testing. [2] No specific treatment is available; however, diet therapy may reduce clinical manifestations.
Signs and symptoms
Symptoms usually first occur during childhood and include the following:
-
Exercise intolerance (premature fatigability)
-
Weakness and stiffness with exercise
-
Painful muscle cramps
See Presentation for more detail.
Diagnosis
Laboratory studies
The following laboratory studies are recommended:
-
Measurement of creatine kinase level
-
Fasting glucose testing
-
Urine studies
-
Complete blood cell count
-
Liver function tests
Other studies
The following tests and procedures are also recommended:
-
Ischemic forearm test
-
Electromyography
-
Muscle biopsy
See Workup for more detail.
Management
Although no specific treatment exists for GSD VII, diet therapy is helpful in some cases. A high-protein diet may help increase muscle function in patients with weakness or exercise intolerance.
See Treatment for more detail.
Background
Glycogen is a complex multibranched polysaccharide that acts as energy storage in the human body and is formed mainly in the liver and skeletal muscles. It plays an important role as a glucose-manipulating system that maintains appropriate glucose levels in the human body. It is degraded to glucose molecules when the body needs energy or is in a "fasting state." On the other hand, it is formed and accumulated in the liver and skeletal muscle when blood glucose is elevated after food intake or during the "fed state."
Glycogen storage disease (GSD) is the result of an enzyme defect. These enzymes normally catalyze reactions that ultimately convert glycogen compounds to glucose. Enzyme deficiency results in glycogen accumulation in tissues, especially liver and skeletal muscle cells. In many cases, the defect has systemic consequences, but in some cases, the defect is limited to specific tissues. Most patients experience muscle symptoms, such as weakness and cramps, although certain GSDs manifest as specific syndromes, such as hypoglycemic seizures or cardiomegaly, based on which enzyme is affected in the carbohydrate metabolic pathway. Each GSD represents a specific enzyme defect, and each enzyme is also specific for certain body cells.
The following list contains a quick reference for 8 of the GSD types:
-
0 - Glycogen synthase deficiency
-
Ia - Glucose-6-phosphatase deficiency (von Gierke disease)
-
II - Acid maltase deficiency (Pompe disease)
-
III - Debranching enzyme deficiency (Forbes-Cori disease)
-
IV - Transglucosidase deficiency (Andersen disease, amylopectinosis)
-
V - Myophosphorylase deficiency (McArdle disease)
-
VI - Phosphorylase deficiency (Hers disease)
-
VII - Phosphofructokinase deficiency (Tarui disease)
The figure below demonstrates where various forms of GSD affect the metabolic carbohydrate pathways.
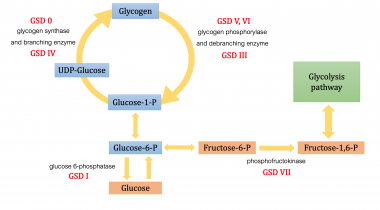 Metabolic pathways of carbohydrate metabolism
Metabolic pathways of carbohydrate metabolism
Although at least 14 unique GSDs are discussed in the literature, the 4 that cause clinically significant muscle weakness are Pompe disease (GSD type II, acid maltase deficiency), Cori disease (GSD type III, debranching enzyme deficiency), McArdle disease (GSD type V, myophosphorylase deficiency), and Tarui disease (GSD type VII, phosphofructokinase deficiency). One form, von Gierke disease (GSD type Ia, glucose-6-phosphatase deficiency), causes clinically significant end-organ disease with significant morbidity. The remaining GSDs are not benign but are less clinically significant; therefore, the physician should consider the aforementioned GSDs when initially entertaining the diagnosis of a GSD. Interestingly, GSD type 0 also is described, which is due to defective glycogen synthase.
These inherited enzyme defects usually present in childhood, although some, such as McArdle disease and Pompe disease, have separate adult-onset forms. [1] In general, GSDs are inherited as autosomal recessive conditions. [3] Several different mutations have been reported for each disorder.
Unfortunately, no specific treatment or cure exists, although diet therapy may be highly effective at reducing clinical manifestations. In some cases, liver transplantation may abolish biochemical abnormalities. Active research continues.
Diagnosis depends on findings from muscle biopsy, electromyography, ischemic forearm testing, creatine kinase testing, patient history, and physical examination. [2] Biochemical assay for enzyme activity is the method of definitive diagnosis.
GSD type VII, or Tarui disease, is named after Dr Seiichiro Tarui, the Japanese physician who first described this disorder in 1965. [4] He found that 3 Japanese siblings whose parents were first cousins had muscle weakness after prolonged exertion. All of them had absent activity of phosphofructokinase (PFK) in the skeletal muscle.
This article will focus mainly on GSD type VII, or PFK deficiency. We will review the pathophysiology, genetics, disease manifestations, diagnosis, and treatment.
Pathophysiology
GSD VII, or Tarui disease, is an autosomal recessive condition caused by deficiency of PFK, the enzyme that catalyzes the rate-limiting step in glycolysis. It turns fructose-6-phosphate (F-6-P) to fructose-1,6-bisphosphate. Enzyme deficiency results in glycolysis blockage and accumulation of F-6-P, which can turn to glucose-6-phosphate (G-6-P), leading to increased glycogen formation. This causes myopathy, including muscle pain and exercise-induced fatigue and weakness. Tarui disease resolves with rest and, although no specific treatment exists, the condition may not progress to severe disability.
Garcia et al investigated the effects of PFK deficiency in tissue other than skeletal muscle on the pathogenesis of GSD type VII. [5] In a study of PFK-deficient mice, the authors found that because the animals' erythrocytes retained only 50% of their PFK activity, severe hemolysis, significant decreases in 2,3-bisphosphoglycerate levels (impairing the extraction of oxygen from hemoglobin), and compensatory reticulocytosis and splenomegaly occurred. Reduced levels of cardiac PFK activity were found as well, which combined with the other hematologic changes, led to the development of cardiac hypertrophy.
Currently, it is known that PFK consists of 3 subunit isozymes including M (muscle), L (liver), and P (platelet). Active PFK has tetrameric structure, and each tissue in the human body has a specific different subunit composition. For example, skeletal myocytes express only M subunit, erythrocytes express both M and L subunit equally, and hepatocytes and nephrons express L subunit in the majority. [6, 7] For GSD type VII, M isozyme is affected. Therefore, muscle cells and red bood cells are affected in patients who have this disease.
Early onset of hyperuricemia and gouty arthritis is found in some patients. It is believed that this condition results from increased shunting of F-6-P to the pentose phosphate pathway, which results in increased production of nucleotide, leading to an increase in uric acid formation and gout crystal formation eventually. Vora et al reported 3 patients with PFK deficiency and hyperuricemia, who had elevated levels of hexose monophosphate. [8]
Some aspects of GSD type VII still have no explanation. For example, brain neurons mainly express M subunit, but there are few data about neurologic involvement in this disease. Madhoun et al reported a unique case of a man with PFK deficiency who also presented with portal and mesenteric vein thrombosis, which did not have clear pathogenesis. [9]
Causes and genetics
GSD type VII is a genetic disorder with autosomal recessive inheritance, caused by homozygous or compound heterozygous mutations in the gene encoding for the M subunit of the PFK enzyme on chromosome 12q13, which contains a total of 24 exons.
Various mutations have been discovered, including missense and frameshift types. Patients of Ashkenazi Jewish descent have been found to have specific splice site mutations in exon 5 and exon 22. [10]
In a study of 5 patients with muscle PFK deficiency from different regions of Italy, Musumeci et al found 4 novel genetic mutations. [11]
Epidemiology
GSDs are very rare conditions. In Europe, Canada, and the United States, the total incidence is between 1 in 20,000 and 1 in 40,000. GSD type VII is extremely rare, and most data are obtained from case reports or case series. The true incidence of the disease may be higher, as clinical features of the disease are mild and affected patients may not be recognized and diagnosed.
Race- and age-related demographics
Raben et al reported that the disease appears to be prevalent among people of Ashkenazi Jewish descent with mutations of the PFKM gene that cause exon slicing site defect. [12, 10]
In general, GSDs present in childhood. Later onset correlates with a less severe form. Most of the infantile forms have a severe course of disease and multisystem involvement, leading to high mortality. [13, 14, 15]
Prognosis
No cure exists for GSD type VII.
Morbidity/mortality
As in McArdle disease, immediate morbidity arises from exercise intolerance. In 1984, Haller et al found that some patients with PFK deficiency have muscle fatigue more rapidly after a high-carbohydrate meal. [16] Unlike in McArdle disease, Haller el al later found that there is no consistent "second wind phenomenon" in GSD type VII. [17] Second wind phenomenon is described as a decrease in heart rate and an improvement in exercise tolerance after 6-8 minutes of aerobic excercise.
Complications
Tarui disease causes exercise intolerance and mild hemolysis.
Patient Education
As with all genetic diseases, genetic counseling is appropriate.
The following patient education resources are available from WebMD:
-
Metabolic pathways of carbohydrate metabolism









