Practice Essentials
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is a hereditary condition resulting from a structural defect in G6PD, a "housekeeping" enzyme that is particularly important for the survival of red blood cells and their ability to respond to oxidative stress. [1] G6PD deficiency is the most common enzyme deficiency in humans, affecting about 400 million people worldwide, with a high prevalence in persons of African, Asian, and Mediterranean descent. [2] It is inherited as an X-linked recessive disorder, and thus most often affects males. G6PD deficiency is polymorphic, with more than 300 variants.
G6PD deficiency confers partial protection against malaria, and geographic prevalence of the disorder correlates with the historical distribution of malaria. This probably accounts for the persistence and high frequency of the responsible genes. [3, 4, 5, 6, 7, 8]
Signs and symptoms of G6PD deficiency
Most patients with G6PD deficiency are asymptomatic. Clinical manifestations may include the following:
-
Neonatal jaundice
-
Episodes of intravascular hemolysis and consequent anemia, triggered by infections, medicines that induce oxidative stresses, fava beans, and ketoacidosis. Hemolysis begins 24 to 72 hours after exposure to oxidant stress. Patients with severe hemolysis present with weakness, tachycardia, jaundice, and hematuria.
-
Chronic hemolytic anemia
See Presentation
Workup in G6PD deficiency
Tests to diagnose hemolysis include the following:
-
Complete blood cell count (CBC) and reticulocyte count
-
Lactate dehydrogenase (LDH) level
-
Indirect and direct bilirubin level
-
Serum haptoglobin level
-
Urinalysis for hematuria
-
Urinary hemosiderin
-
Peripheral blood smear (with Heinz body prep)
Tests for G6PD deficiency include the following:
-
Semi-quantitative tests -The fluorescent spot test (not reliable in females)
-
Quantitative tests (spectrophotometric) - the criterion standard.
-
Point-of-care tests – Newer versions are quantitative; potential for use in both males and females, in basic clinical laboratories in both high- and low-resource settings
See Workup
Management
Most individuals with G6PD deficiency do not require any treatment. Acute hemolytic anemia in G6PD-deficient patients is largely preventable by avoiding exposure to fava beans, drugs, and chemicals that can cause oxidant stress. Identification and discontinuation of the precipitating agent is critical in the management of hemolysis in patients with G6PD deficiency.
Acute hemolysis is usually self-limiting, often resolving after 8 to 14 days. Rarely, transfusion is needed in cases of severe anemia.
Infants with prolonged neonatal jaundice as a result of G6PD deficiency should receive phototherapy. Exchange transfusion may be necessary in cases of severe neonatal jaundice.
Persons with chronic hemolysis or nonspherocytic anemia should be placed on daily folic acid supplements. Consultations with a hematologist are ideal for long-term follow up
See Treatment.
Pathophysiology
The G6PD enzyme is part of the pentose monophosphate shunt. It catalyzes the oxidation of glucose-6-phosphate and the reduction of nicotinamide adenine dinucleotide phosphate (NADP+) to nicotinamide adenine dinucleotide phosphate (NADPH). NADPH maintains glutathione in its reduced form, which acts as a scavenger for dangerous oxidative metabolites.
The pentose monophosphate shunt is the only source for NADPH in red blood cells. Therefore, red blood cells depend on G6PD activity to generate NADPH for protection. Thus, red blood cells are more susceptible to oxidative stresses than other cells. In persons with G6PD deficiency, oxidative stresses can denature hemoglobin and cause intravascular hemolysis. Denatured hemoglobin can be visualized as Heinz bodies in peripheral blood smears processed with supravital staining. Heinz bodies are shown in the figure below.
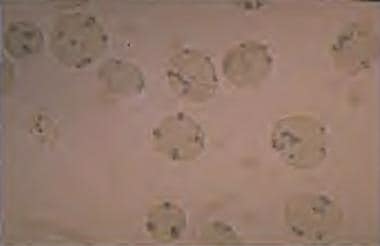 G6PD deficiency: Heinz bodies in a peripheral smear stained with a supravital stain. Heinz bodies are denatured hemoglobin, which occurs in G6PD deficiencies and in unstable hemoglobin disorders.
G6PD deficiency: Heinz bodies in a peripheral smear stained with a supravital stain. Heinz bodies are denatured hemoglobin, which occurs in G6PD deficiencies and in unstable hemoglobin disorders.
The degree of G6PD deficiency determines the clinical expression of the disorder. Individuals with minimally reduced enzyme levels do not experience hemolysis. Others with a greater degree of deficiency have episodes of brisk hemolysis triggered by infections, taking drugs that increase oxidative stress, ingesting fava beans, or ketoacidosis. Hemolysis due to oxidant stresses are usually self-limiting within 8 to 14 days due to the compensatory production of young red blood cells with high levels of G6PD. Patients with severe G6PD deficiency have chronic hemolysis and are often thought to have non-spherocytic hemolytic anemia.
Jaundice in G6PD-deficient neonates is considered to be due to an imbalance between the production and conjugation of bilirubin, with a tendency for inefficient bilirubin conjugation. Borderline premature infants are at special risk of the bilirubin production-conjugation imbalance. [9]
Etiology
The gene that codes for G6PD is located in the distal long arm of the X chromosome at the Xq28 locus. The G6PD gene is 18 kilobases (kb) long with 13 exons, and the G6PD enzyme has 515 amino acids. Currently, 186 mutations in the G6PD gene have been documented. Most are single-base changes that result in an amino acid substitution. [7]
G6PD deficiency is an X-linked recessive disorder, with an inheritance pattern similar to that of hemophilia and color blindness: males usually manifest the abnormality and females are carriers. Females may be symptomatic if they are homozygous or if inactivation of their normal X chromosome occurs. The allele for G6PD has been used to establish clonality. [6, 7]
Specific G6PD alleles are associated with G6PD variants with different enzyme levels and, thus, different degrees of clinical disease severity. The variation in G6PD levels accounts for differences in sensitivity to oxidants. Chronic hemolysis occurs with extremely low enzyme levels.
The G6PD A+ variant is associated with high enzyme levels and, hence, no hemolysis. G6PD A- is associated with lower enzyme levels and acute intermittent hemolysis. G6PD A- occurs in high frequency in African, Mediterranean, and Asian variants. Mediterranean G6PD A- (also called G6PD Mediterranean) is characterized by enzyme deficiencies that are more severe than in the other G6PD A- alleles. Fava bean hemolysis usually occurs in Mediterranean G6PD deficiency disorders. G6PD B is the wild type of allele (normal variant).
The World Health Organization has classified the different G6PD variants according to the degree of enzyme deficiency and severity of hemolysis, into classes I-V. Class I deficiencies are the most severe. G6PD Mediterranean deficiency usually is a class II deficiency and G6PD A- deficiency is a class III deficiency. Classes IV and V are of no clinical significance. [6, 7]
Epidemiology
Glucose-6-phosphatase dehydrogenase (G6PD) deficiency occurs worldwide. In the United States, Black males are primarily affected, with a prevalence of about 10%. Internationally, the geographic prevalence of the disorder correlates with the distribution of malaria. The highest prevalence rates (with gene frequencies from 5-25%) are found in the following regions [10, 11, 2] :
-
Tropical Africa
-
The Middle East
-
Tropical and subtropical Asia
-
Some areas of the Mediterranean
-
Papua New Guinea
Mortality/morbidity
Most persons with G6PD deficiency are asymptomatic. Symptomatic patients can present with neonatal jaundice and acute hemolytic anemia. [6, 7]
Kernicterus is a rare complication of neonatal jaundice, [12] but can occur in certain populations and can be fatal. Other mechanisms may contribute to hyperbilirubinemia in G6PD deficiency, such as an underlying defect in uridine diphosphoglucoronate-glucuronosyltransferase, the enzyme affected in Gilbert syndrome (see Unconjugated Hyperbilirubinemia).
Acute episodic hemolytic anemia can occur due to oxidant stress induced by exposure to certain drugs or chemicals (including some anesthetic agents [13] ), infections, ketoacidosis, or the ingestion of fava beans. [10, 14, 15, 16] Chronic hemolysis occurs in severe G6PD deficiency. Fatality rarely occurs.
G6PD deficiency appears to be a risk factor for the development of diabetes mellitus. A systematic review and meta-analysis by Lai and colleagues of publications involving involving 949,260 persons with G6PD deficiency found an odds ratio (OR) of 2.37 (95% confidence interval 1.50-3.73) for diabetes. The risk was higher in men than in women (OR 2.22 versus 1.87, respectively). [17] Certain G6PD gene variants in Africans and East Asians (G6PD-Asahi, G6PD-Canton, G6PD-Kaiping) have been shown to lower glycosylated hemoglobin (HbA1c) levels independent of glycemia, so patients who are carriers of those variants may be at risk for underdiagnosis of diabetes or pre-diabetes if screened by HbA1c without confirmation by direct glucose measurements. [18]
Individuals with G6PD deficiency are at increased risk for severe COVID-19 infection and long COVID. [19, 20] However, G6PD deficiency may have a protective effect on ischemic heart disease, cerebrovascular disease, and colorectal cancer. [21, 22]
Racial and sexual disparities
G6PD deficiency affects all races, but the highest prevalence is in persons of African, Asian, or Mediterranean descent. [10, 14] In the US, G6PD deficiency is prevalent in approximately 14% of males of African descent. [23]
The severity of G6PD deficiency varies significantly among racial groups. Variants producing severe deficiency primarily occur in the Mediterranean population. African populations have milder hemolysis due to higher enzyme levels.
G6PD deficiency is an X-linked inherited disease that primarily affects men. Women may be affected if they are homozygous, which occurs in populations in which the frequency of G6PD deficiency is quite high. Heterozygous women (carriers) can experience clinical disease as a result of X chromosome inactivation, gene mosaicism, or hemizygosity. [24]
-
G6PD deficiency: Heinz bodies in a peripheral smear stained with a supravital stain. Heinz bodies are denatured hemoglobin, which occurs in G6PD deficiencies and in unstable hemoglobin disorders.



