Practice Essentials
Diffuse proliferative glomerulonephritis (DPGN) is a term used to describe a distinct histologic form of glomerulonephritis common to various types of systemic inflammatory diseases, including autoimmune disorders (eg, systemic lupus erythematosus [SLE]), vasculitis syndromes (eg, granulomatosis with polyangiitis), and infectious processes. In DPGN, more than 50% of the glomeruli (diffuse) show an increase in mesangial, epithelial, endothelial (proliferative), and inflammatory cells (ie, glomerulonephritis). (Increased cellularity is demonstrated in the image below.)
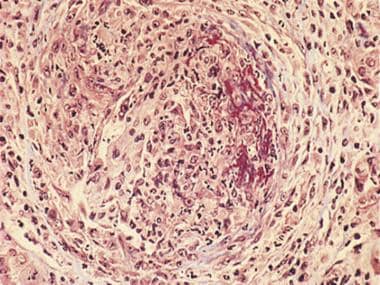 Light microscopy (trichrome stain) shows globally increased cellularity, numerous polymorphonuclear cells, cellular crescent (at left of photomicrograph) and fibrinoid necrosis (brick red staining at right of photomicrograph). These findings are characteristic of diffuse proliferative glomerulonephritis.
Light microscopy (trichrome stain) shows globally increased cellularity, numerous polymorphonuclear cells, cellular crescent (at left of photomicrograph) and fibrinoid necrosis (brick red staining at right of photomicrograph). These findings are characteristic of diffuse proliferative glomerulonephritis.
In contrast, when fewer than 50% of the glomeruli are involved, the condition is termed focal proliferative glomerulonephritis. However, this entity has the potential to progress to DPGN.
The diagnosis of DPGN is often suspected in a patient presenting with a systemic inflammatory disease who manifests hematuria, proteinuria, and active urinary sediment or azotemia (ie, rise in serum urea nitrogen, creatinine). Histologic findings from kidney biopsy tissue are used to confirm the diagnosis.
Sporadic forms of kidney diseases that can manifest histologically as focal, segmental, necrotizing, and crescentic glomerulonephritis or DPGN with undetermined incidence include the following:
-
Microscopic polyangiitis
-
Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome)
-
Essential mixed cryoglobulinemia, which also may manifest as membranoproliferative glomerulonephritis
-
Henoch-Schönlein purpura
-
Connective tissue diseases
In severe forms of DPGN, epithelial proliferation obliterates the Bowman space (ie, crescents). The resulting acute kidney injury may manifest as an acute anuria or a steady decline in kidney function. Spontaneous remission is rare, and treatment results are anecdotal.
Pathophysiology
Most cases of DPGN result from the deposition of immune complexes in the mesangium, glomerular basement membrane (GBM), subendothelial or subepithelial locations. Antibodies may form immune complexes with circulating antigens before deposition (ie, immune complex deposition) or may bind directly to nonglomerular antigens already present in the mesangium or GBM (ie, in situ immune complex formation). In anti-GBM disease, the antibodies act against the GBM. The pathogenesis of antineutrophil cytoplasmic antibody (ANCA)–associated glomerulonephritis is unknown, although microvasculitis is the predominant feature without immune complex formation.
Activation of the complement system through the classic pathway by immune complexes or direct cell-mediated injury in ANCA-associated glomerulonephritis results in the recruitment of inflammatory cellular infiltrates (eg, lymphocytes, macrophages, neutrophils), proliferation of the mesangial and endothelial cells, and necrosis. Cellular crescents and fibrin thrombi may be present in more severe cases. The net result is obliteration of the capillary loops and sclerosis, predisposing the patient to hypertension and renal failure.
The cellular and immunologic attack on the glomerulus renders the GBM permeable to protein, red blood cells (RBCs), and white blood cells (WBCs). Therefore, urinalysis during active inflammation (or glomerulonephritis) characteristically shows an active urinary sediment, with RBCs or casts, WBCs or casts, and variable degrees of proteinuria (ie, nephritic pattern).
Anti-GBM disease is an autoimmune disease in which autoantibodies are directed against type IV collagen in the GBM. Binding of these autoantibodies to the GBM induces rapidly progressive glomerulonephritis (RPGN) and crescentic glomerulonephritis. The clinical complex of anti-GBM nephritis and lung hemorrhage is Goodpasture syndrome. The typical morphologic pattern seen on light microscopy is DPGN, with focal necrotizing lesions and crescents in more than 50% of glomeruli (ie, crescentic glomerulonephritis). Acute nephrotic syndrome is rare, and a bimodal peak in incidence exists. Although any age group may be affected, the first peak in incidence occurs in the third to the sixth decades of life and the second occurs in the sixth to the seventh decades of life.
In patients with granulomatosis with polyangiitis (GPA; previously known as Wegener granulomatosis), kidney biopsy findings typically reveal focal, segmental, necrotizing, pauci-immune glomerulonephritis with crescent formation.
In microscopic polyangiitis (MPA), the usual histopathologic lesion is a pauci-immune focal segmental necrotizing and crescentic glomerulonephritis. In eosinophilic granulomatosis with polyangiitis, a minority of patients may develop focal segmental necrotizing glomerulonephritis; in mixed cryoglobulinemias, the characteristic morphologic lesions are diffuse mesangial proliferative or membranoproliferative glomerulonephritis. For Henoch-Schönlein purpura, light microscopic appearances can vary from mild mesangial proliferation and expansion to diffuse proliferation with glomerular crescents.
In rheumatoid arthritis (RA), lesions of mesangial proliferative glomerulonephritis and basement membrane thickening caused by subepithelial immune deposits may be observed. Occasional cases of focal mesangial proliferative glomerulonephritis with mesangial deposition of immunoglobulin G (IgG) and complement have been described in polymyositis and dermatomyositis. [1]
In addition to poststreptococcal glomerulonephritis, nephritic syndrome and RPGN can complicate acute immune-complex glomerulonephritis due to other viral, bacterial, fungal, and parasitic infections. Some of these warrant specific mention. Diffuse proliferative immune complex glomerulonephritis is a well-described complication of acute and subacute bacterial endocarditis and usually is associated with hypocomplementemia. The glomerular lesion typically resolves following eradication of the cardiac infection. Shunt nephritis is a syndrome characterized by immune complex glomerulonephritis secondary to infection of ventriculoatrial shunts inserted for treatment of childhood hydrocephalus.
The most common offending organism is coagulase-negative Staphylococcus. Renal impairment usually is mild and is associated with hypocomplementemia. Nephrotic syndrome complicates 30% of cases.
Acute proliferative glomerulonephritis can also complicate chronic suppurative infections and visceral abscesses. Patients typically present with a fever of unknown origin and an active sediment. Although renal biopsy is used to detect immune deposits containing IgG and C3, serum complement levels usually are within the reference range.
Etiology
Systematic diseases causing DPGN include the following:
-
Lupus nephritis class IV
-
IgA nephropathy
-
Goodpasture syndrome
-
Granulomatosis with polyangiitis (GPA)
-
Microscopic polyangiitis MPA)
-
Henoch-Schönlein purpura
-
Cryoglobulinemia
-
Vasculitis
Immunoglobulin A (IgA) nephropathy and SLE are the most common etiologies. In patients with lupus nephritis class IV, histologic transformation from one class to another is recorded in up to 40% of repeat biopsies. The most likely transformation is from class II or III to class IV. Any other class may be superimposed on class V.
GPA (formerly known as Wegener granulomatosis), MPA, and eosinophilic granulomatosis with polyangiitis (EGPA, Churg-Strauss syndrome) are termed antineutrophil cytoplasmic antibody (ANCA)–associated vasculitides (AAVs). [2] GPA, MPA, and EGPA are also termed pauci-immune glomerulonephritis and are characterised by necrotizing inflammation of the small vessels: arterioles, capillaries, and venules with little or no deposition of immune complexes in the vessel wall (pauci-immune). About 10% of patients presenting with MAP, GPA, and EGPA are ANCA negative. [3]
Post-infectious glomerulonephritis may occur in association with bacterial, viral, fungal, protozoal, and helminthic infection but is most often secondary to streptococcal sore throat or skin infection and occurs 2-4 weeks after infection. Other common infectious causes of DPGN are infective endocarditis, hepatitis B, and hepatitis C. Shunt nephritis is an immune complex–mediated glomerulonephritis that develops as a complication of chronic infection on ventriculoatrial or ventriculojugular shunts inserted for the treatment of hydrocephalus [4]
COVID-19 has been associated with a cluster of of 8 cases of anti-GBM disease. [5]
Epidemiology
Frequency
United States
The reported prevalence of kidney disease in systemic lupus erythematosus is about 38%, and the incidence of end-stage renal disease (ESRD) attributed to lupus nephritis in adults is 4.5 cases per million in the general population. [6]
IgA nephropathy accounts for about 10% of biopsies performed for glomerular disease in the United States. Prevalence rates are lower in the United States than in Asian countries.
Anti–glomerular basement membrane (GBM) disease is a rare disorder of unknown etiology with an annual incidence of 0.5-1 case per million. [7] About 50-70% of patients have lung hemorrhage; anti-GBM antibodies develop in the serum of more than 90% of patients with anti-GBM nephritis, according to findings on specific radioimmunoassay.
Cytoplasmic antineutrophil cytoplasmic antibodies (ANCAs) are detected at presentation in 80% of patients with renal disease and in 10% more during follow-up. In contrast with ANCA-associated disease involving the lung, granulomas rarely develop in the kidney.
Most cases of acute poststreptococcal glomerulonephritis are sporadic, although the disease can occur as an epidemic. The characteristic lesion visible on light microscopy is DPGN. Crescents may be present, and extraglomerular involvement usually is mild.
Nephritis is present in 80% of cases of Henoch-Schönlein purpura and manifests as a nephrotic urine sediment and moderate proteinuria. Macroscopic hematuria and nephrotic range proteinuria are uncommon.
International
The incidence of DPGN in kidney biopsies varies from approximately 10-27% in Europe and 30% in the Middle East to 41% in Japan. The most common glomerulopathy is due to immunoglobulin A (IgA) nephropathy.
Distribution of IgA nephropathy varies in different geographic regions throughout the world. High prevalence rates are observed in Singapore, Japan, Australia, Hong Kong, Finland, and southern Europe, whereas low prevalence rates are the rule in the United Kingdom, Canada, and the United States. IgA nephropathy is observed in up to 40% of all biopsies performed for glomerular disease in Asia, compared with 20% in Europe and 10% in North America.
Worldwide, up to 80% of patients with Henoch-Schönlein purpura (ie, anaphylactoid purpura), which is a distinct systemic vasculitis syndrome that is characterized by palpable purpura (most commonly distributed over the buttocks and lower extremities), arthralgias, and gastrointestinal signs and symptoms, have DPGN.
The prevalence of AAV in Europe is estimated at to be 46–184 per million with annual incidence rates per million estimated at 2.1–14.4 for GPA, 2.4–10.1 for MPA and 0.5–3.7 EGPA. [2]
Race
Lupus nephritis differs with ethnicity. Whites (12–33%) are less likely to have lupus nephritis than Blacks (40–69%), Hispanics (36–61%), or Asians (47–53%). [6] Additionally, Black and Hispanic patients with SLE develop lupus nephritis earlier than White patients and have worse outcomes, including death and ESRD, [8]
Sex
Men tend to have more aggressive disease than women. However, for SLE, the female-to-male incidence ratio is 9:1 for women of childbearing age. By comparison, the female-to-male ratio is only 2:1 for disease developing during childhood or in people aged 65 or older. Males who develop SLE have the same incidence of renal disease as do females. [9]
Microscopic PAN is more common in males (ie, male-to-female ratio of 2:1). The distribution of granulomatosis with polyangiitis among the sexes is roughly equal, with a slight male predominance. Males have a 2.7 times higher incidence of IgA nephropathy than females.
Age
Patients with Goodpasture syndrome typically are young males aged 5-40 years (the male-to-female ratio is 6:1). In contrast, patients presenting during the second peak in incidence, occurring in the sixth decade of life, rarely experience lung hemorrhage and have an almost equal sex distribution.
SLE occurs in all age groups, with the peak incidence occurring in women of childbearing age. Over 85% of patients are younger than 55 years.
Granulomatosis with polyangiitis develops in people of any age. Approximately 15% of patients are younger than 19 years, and only rarely does the disease occur before adolescence. The mean age of onset is approximately 40 years. The mean age of patients at onset in reports of PAN and microscopic polyangiitis is 48 years.
Prognosis
Evidence of glomerulosclerosis, fibrous crescents, tubular atrophy, and, particularly, interstitial fibrosis on light microscopy indicates advanced disease and a poor prognosis. Males are at a higher risk factor for a poor prognosis. [9] Other risk factors associated with a poor prognosis include heavy proteinuria, hypertension, interstitial fibrosis, oliguria, and azotemia at presentation. Overall, about 50% of patients with DPGN require dialysis within 6-12 months after presentation.
About 10% of acute kidney injuries in adults are attributed to glomerulonephritis mainly associated with granulomatous polyangiitis (GPA), microscopic polyangiitis (MPA), and anti–glomerular basement membrane (GBM) disease. The prognosis improves with early diagnosis and treatment. [10]
Renal survival is best with IgA nephropathy, which has an indolent course with a favorable outcome. Death due to DPGN in IgA nephropathy is rare.
Patient and kidney survival in untreated anti-GBM disease is poor. Patient and kidney survival have shown improvement with treatment, but disease progression can be very rapid, and outcome is related to the severity at presentation. A retrospective analysis of 123 patients with anti-GBM glomerulonephritis seen at six centers between 1986 and 2015 reported a 5-year kidney survival rate of 34%. However, the success rate improved significantly in patients diagnosed after 2007. Dialysis dependency, low percentage of normal glomeruli, and large extent of interstitial infiltrate were associated with poor kidney outcome in this series. [11]
Lupis nephritis (LN) is a major risk factor for morbidity and mortality in SLE; 10% of patients with LN will develop ESRD. [8] In some series, the rate of progression to ESRD in class IV lupus nephritis was 50% during a 2-year follow-up. [12, 9, 13, 14] Patients with LN also have a higher standardized mortality ratio (6–6.8 versus 2.4) and die earlier than SLE patients without LN. However, 10-year survival improves from 46% to 95% if disease remission can be achieved. [8]
Advances in immunosuppressive therapy and renal replacement therapy have markedly reduced the mortality and morbidity rates of DPGN in the last 2 decades. A significant portion of the morbidity and mortality in DPGN is due to complications of immunosuppressive therapy, including drug toxicity and infection.
Treatment has dramatically improved the prognosis of patients diagnosed with ANCA-associated vasculitis (AAV). The median survival from diagnosis without treatment is 5 months, whereas 88% of treated patients survive 1-year and 78% survive for 5 years. Almost 50% of deaths are related to infection and 20% from disease-related complications in the first year. In subsequent years, cardiovascular disease and malignancy are the major causes of mortality, followed by infectious (20%) and vasculitis-specific (6%) causes. Severe renal involvement and diffuse alveolar hemorrhage are the major causes of vasculitis-related mortality. [15]
Mortality due to poststreptococcal glomerulonephritis is rare. Prior to the introduction of immunosuppressive therapy, more than 80% of patients with anti-GBM nephritis developed ESRD within 1 year, and many patients died from pulmonary hemorrhage or complications of uremia and infection. However, in a study of atypical anti-GBM nephritis characterized by an indolent course, no pulmonary involvement, and undetectable circulating α3NC1 antibodies, the 1-year patient and renal survival rates were 93% and 85%, respectively. [16]
Patient Education
Educate patients on the disease process, renal prognosis, complications of therapy, and importance of adhering to the treatment plan. The importance of keeping appointments must be emphasized. For those with advanced kidney failure, options for renal replacement therapy (ie, hemodialysis, peritoneal dialysis, transplantation) should be fully discussed.
For patient education information, see Blood in the Urine.
For further information, see Mayo Clinic - Kidney Transplant.
-
Light microscopy (trichrome stain) shows globally increased cellularity, numerous polymorphonuclear cells, cellular crescent (at left of photomicrograph) and fibrinoid necrosis (brick red staining at right of photomicrograph). These findings are characteristic of diffuse proliferative glomerulonephritis.
-
Diffuse proliferative glomerulonephritis (DPGN). Immunofluorescent microscopy shows (except for anti–glomerular basement membrane [GBM] disease) a granular deposition of immunoglobulins, complement, and fibrin along the GBM, tubular basement membranes, and peritubular capillaries (image 2a). Linear deposition occurs in the GBM in anti-GBM disease (image 2b).
-
Diffuse proliferative glomerulonephritis (DPGN). Using electron microscopy, electron-dense deposits are visible in the mesangial, subendothelial, intramembranous, and subepithelial locations.