Practice Essentials
Hereditary elliptocytosis (HE) is a group of disorders of the red blood cell (RBC) membrane that are characterized by elliptical-shaped erythrocytes (elliptocytes; see the image below) and shortened RBC survival. Unlike normal RBCs, which repeatedly and momentarily assume an elliptical shape to negotiate through capillaries but then regain their biconcave discoid shape after they pass through the microcirculation, the RBCs in HE lack the elastic recoil necessary for returning to the discoid shape and eventually assume the fixed characteristic morphology of elliptocytes, with a decreased surface-to-volume ratio. These elliptocytes are not as deformable as normal RBCs and are eventually trapped and removed by the spleen; this manifests as hemolytic anemia. [1, 2, 3, 4]
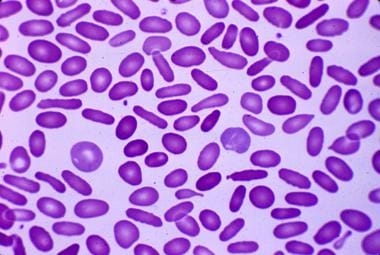 Hereditary elliptocytosis: Peripheral blood smear reveals cigar-shaped erythrocytes (elliptocytes). Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
Hereditary elliptocytosis: Peripheral blood smear reveals cigar-shaped erythrocytes (elliptocytes). Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
HE is due to defects in either the structure or quantity of the cytoskeletal proteins responsible for maintaining the biconcave morphology of RBCs. Mutations in either alpha- and beta-spectrin are most commonly responsible, but mutations in other cytoskeletal proteins (band 4.1 and glycophorin) are also described. [5] Most of these disorders are clinically silent, with only some forms associated with clinically significant hemolysis.
The mode of inheritance is autosomal dominant, except for hereditary pyropoikilocytosis (HPP), which is autosomal recessive. Instances of spontaneous mutations are rare.
In most cases, HE causes no symptoms and requires no therapy. For patients with clinically significant hemolytic anemia, splenectomy provides marked improvement. See Treatment.
See also Pediatric Hereditary Elliptocytosis and Related Disorders.
Pathophysiology
HE results from defects in the protein scaffolding of the erythrocyte membrane, which decrease the deformability and resilience of the RBCs. Normal RBCs are 7 microns in diameter and assume the shape of a biconcave disk with central pallor. They are rugged cells and can survive in the circulation for 120 days as they repeatedly and momentarily assume an elliptical shape to negotiate through capillaries as small as 2-3 microns in diameter.
After passing through the microcirculation, normal RBCs can regain their discoid shape because of their elastic recoil; however, the RBCs in HE fail to do so. This failure to regain their discoid shape eventually produces the fixed characteristic morphology of elliptocytes with a decreased surface-to-volume ratio. These elliptocytes are not as deformable as normal RBCs and are eventually trapped and removed by the spleen. This process of premature destruction (ie, cells surviving < 120 d) is the basis of the extravascular hemolysis that clinically defines these disorders.
The RBC membrane is composed of a fragile lipid bilayer stretched over a flexible protein cytoskeleton. Spectrin is the major component of this scaffold and consists of 2 chains, alpha and beta, which are encoded by separate genes and which are twisted together to form an elongated heterodimer. At the head region, the heterodimers associate to form tetramers. At the distal end, they bind to other cytoskeletal proteins, namely actin and protein 4.1. These proteins, in turn, anchor the scaffold to the lipid bilayer by linking to the transmembrane proteins band 3, glycophorin A, and glycophorin C.
Mutations in either of the spectrins, glycophorin C, or band 4.1 account for most cases of HE. [6] Different point mutations are described in various families and account for some the clinical variability of this disorder. Mutations affecting the level (but not the structure) of glycophorin C (Leach phenotype) are also described. These mutations collectively result in defective assembly of the protein scaffolding on the inner aspect of the RBC membrane. The most common group of mutations affect alpha- or beta-spectrin and result in defects in the formation of the spectrin heterodimer or in the association of the heterodimer with the lipid anchoring complex (formed by actin, band 3, protein 4.1 and glycophorin C). [7]
Taken together, all of these defects result in defects in membrane stability and deformability as the RBCs pass through the microcirculation. The spleen removes the damaged erythrocytes, diminishing erythrocyte survival. Therefore, as with other chronic hemolytic disorders, clinical sequelae of HE may include splenomegaly and a propensity to develop gallstones, along with a variable degree of anemia.
Etiology
Hereditary elliptocytosis (HE) and hereditary pyropoikilocytosis (HPP) are heterogeneous red blood cell (RBC) membrane disorders that result from mutations in the genes encoding α-spectrin (SPTA1), β-spectrin (SPTB), or protein 4.1R (EPB41). HE is caused by monoallelic (heterozygous) mutations and inherited in an autosomal dominant fashion, while HPP has an autosomal recessive inheritance and is typically caused by biallelic (homozygous or compound heterozygous) mutations. [8] [9]
Mutations in SPTA1 are the most common, occurring in 65% of HE cases, followed by mutations in SPTB (30%) and EPB41 (5%). [6] HE due to mutations in GYPC causing a complete deficiency of glycophorin C leading to a partial deficiency of protein 4.1R in the junctional complex have been reported. [10]
The clinical phenotypses and associated genes in all RBC membrane disorders are summarized in Table 1. below.
Table 1. RBC Membrane Disorders [6, 2] (Open Table in a new window)
| Phenotype | Gene | Inheritance |
|---|---|---|
| Disorders with RBC membrane structural defect | ||
| HE Type 1 | EPB41 | AD |
| HE Type 2 | SPTA1 | AD |
| HE Type 3 | SPTB | AD |
| HPP | SPTA1 | AR |
| SAO | SLC4A1 | AD |
| HS Type 1 | ANK1 | AD/AR |
| HS Type 2 | SPTB | AD |
| HS Type 3 | SPTA1 | AR |
| HS Type 4 | SLC4A1 | AD |
| HS Type 5 | EPB42 | AR |
| Disorders with altered RBC membrane permeability | ||
| DHS 1 | PIEZO1 | AD |
| DHS 2 | KCNN4 | AD |
| OHS | RHAG | AD |
| CHC | ABCB6 | AD |
| FP | SLC4A1 | AD |
AD: autosomal dominant; AR: autosomal recessive; CHC: cryohydrocytosis; DHS: dehydrated hereditary stomatocytosis; FP: familial pseudohyperkalemia; HE: hereditary elliptocytosis; HPP: hereditary pyropoikilocytosis; HS: hereditary spherocytosis; OHS: overhydrated hereditary stomatocytosis; SAO: Southeast Asian ovalocytosis
|
||
Epidemiology
The worldwide incidence of HE is 1:2000–4000 individuals, but it is as high as 1:100 in some African regions endemic for malaria because of relative resistance of elliptocytes against malaria. [2] In equatorial Africa, the incidence is approximately 0.6%; in Malayan aborigines, the incidence is as high as 30%. However, the true incidence is unknown because many patients do not have any symptoms. [11]
Although no racial or ethnic group is spared, some variants of HE occur more frequently in certain ethnic populations than in others. HE with neonatal poikilocytosis occurs almost exclusively in African-American families, but spherocytic elliptocytosis most commonly affects individuals of European descent. Southeast Asian ovalocytosis (SAO) is a very common condition in the aboriginal peoples from Papua New Guinea, Indonesia, Malaysia, the Philippines, and southern Thailand, in areas where malaria is endemic, with prevalence varying between 5% and 25%. [2]
Because HE is an autosomal disorder, the distribution between the sexes is equal. HE is a congenital disease. However, other acquired disorders, such as myelofibrosis and myelophthisic anemias, may affect the degree of hemolysis.
Prognosis
Most patients with the common form of HE are asymptomatic. Only 5-20% develop uncompensated hemolysis with anemia. Even those with clinically significant hemolysis have an excellent prognosis after splenectomy. Other findings consistent with chronic hemolysis are splenomegaly, pigmented gallstones, leg ulcers, and elevated reticulocyte counts.
Patient Education
Patients should be informed about the autosomal dominant inheritance of the major types of HE. Despite the asymptomatic nature of the disease, family members can be encouraged to be screened for HE.
-
Hereditary elliptocytosis: Peripheral blood smear reveals cigar-shaped erythrocytes (elliptocytes). Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
-
Bizarre RBC morphology seen in hereditary pyropoikilocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
Tables
| Phenotype | Gene | Inheritance |
|---|---|---|
| Disorders with RBC membrane structural defect | ||
| HE Type 1 | EPB41 | AD |
| HE Type 2 | SPTA1 | AD |
| HE Type 3 | SPTB | AD |
| HPP | SPTA1 | AR |
| SAO | SLC4A1 | AD |
| HS Type 1 | ANK1 | AD/AR |
| HS Type 2 | SPTB | AD |
| HS Type 3 | SPTA1 | AR |
| HS Type 4 | SLC4A1 | AD |
| HS Type 5 | EPB42 | AR |
| Disorders with altered RBC membrane permeability | ||
| DHS 1 | PIEZO1 | AD |
| DHS 2 | KCNN4 | AD |
| OHS | RHAG | AD |
| CHC | ABCB6 | AD |
| FP | SLC4A1 | AD |
AD: autosomal dominant; AR: autosomal recessive; CHC: cryohydrocytosis; DHS: dehydrated hereditary stomatocytosis; FP: familial pseudohyperkalemia; HE: hereditary elliptocytosis; HPP: hereditary pyropoikilocytosis; HS: hereditary spherocytosis; OHS: overhydrated hereditary stomatocytosis; SAO: Southeast Asian ovalocytosis
|
||



