Practice Essentials
Cystinuria is an autosomal-recessive defect in the reabsorptive transport of cystine and the dibasic amino acids ornithine, arginine, and lysine from the luminal fluid of the renal proximal tubule and small intestine. The only phenotypic manifestation of cystinuria is cystine urolithiasis, which often recurs throughout an affected individual’s lifetime. [1] See the image below.

Although cystinuria accounts for only about 1-2% of kidney stones in adults, this disorder can result in significant morbidity beginning at a young age, with more frequent stone events and a greater need for surgical intervention than in other urolithiasis disorders, and a potentially faster progression to kidney insufficiency. [2] On average, individuals with untreated cystinuria experience one new stone every year and require a surgical procedure to remove the stones every 3 years. By middle age, the average cystinuria patient will have undergone seven surgical procedures. [3]
The diagnosis of cystinuria is readily made by stone analysis, microscopic examination of the urine, and 24-hour urine testing. Although surgical intervention is necessary for large calculi that are unlikely to pass spontaneously and those that are causing obstruction or symptoms, the cornerstones of treatment are dietary and medical prevention of recurrent stone formation. [4]
For patient education information, see the Kidney Stones Health Center.
Background
In 1810, Wollaston first described a different type of urinary calculus from the urinary bladder and coined the term cystic oxide. [5] Berzelius recognized that the compound was not an oxide, and he named it cystine because the material originated from the bladder. [6]
In 1908, Sir Archibald Garrod identified cystinuria as one of the original "inborn errors of metabolism." [7] Yeh et al [8] and Dent and Rose [9] showed abnormal excretion of the dibasic amino acids lysine, arginine, and ornithine in persons with cystinuria. In 1955, Harris et al reported the complex autosomal-recessive pattern of inheritance of cystinuria. [10] In 1961, Milne et al demonstrated reduced intestinal absorption of dibasic amino acids in people with cystinuria. [11]
In 1954, while studying skin sensitivity to penicillin and its derivatives, Tabachnick et al. noted that one of the degradation products of penicillin, penicillamine, reacted with cystine to form a mixed disulfide, penicillamine cysteine. [12] In 1963, Crawhall et al. first used penicillamine to treat patients with cystinuria. [13]
In recent years, understanding of the genetic and molecular components of cystinuria has advanced. In 1993, Lee et al. cloned a human complementary DNA, rBAT (renal basic amino acid transporter) in chromosome 2, encoding a transport protein for cystine and dibasic amino acids. [14] In 1997, Bisceglia et al. identified type III cystinuria on band 19q13.1. [15]
Pathophysiology
Renal transport of cystine
Amino acids are readily filtered by the glomerulus and undergo nearly complete reabsorption by proximal tubular cells. Only 0.4% of the filtered cystine appears in the urine. Various authors have studied amino acid transport in cell membranes obtained from the proximal renal tubule of humans, rats, and rabbits. At least 2 transport systems are responsible for cystine reabsorption, as follows:
-
High-affinity system: This system is affected in persons with cystinuria. The high-affinity system mediates the uptake of 10% of L-cystine and the dibasic amino acids at the apical membrane of the straight third segment (S3) of the proximal tubule.
-
Low-affinity system: This system is present in the S1-S2 part of the proximal tubule and is responsible for 90% of L-cystine reabsorption. The low-affinity process augments the high-affinity process. After absorption, each molecule of cystine is intracellularly converted to 2 molecules of cysteine. Cysteine exits at the basolateral membrane.
Genetic studies of DNA from families with cystinuria reveal a defective gene located on chromosome 2. The gene that codes for the cystine transporter, initially termed rBAT, is now known as SLC3A1 (SLC for solute carrier) in the International Genome Database. A second cystinuria gene on chromosome 19 is called SLC7A9. [16] The normal SLC7A9 gene encodes a subunit of the cystine transporter called b 0,+ AT (amino acid transporter). The process of cystine uptake is activated by the SLC3A1 and SLC7A9 gene products. Transport of L-cystine in the brush-border membrane vesicle is sodium-independent and electrogenic. In people with cystinuria, the movement of cystine or cysteine from the tubular cells into the blood is not affected. [17]
Intestinal transport of cystine
The high-affinity transporter is present in the apical brush-border membrane of the jejunum and is responsible for absorption of cystine and dibasic amino acids. Most patients with cystinuria have impaired absorption of cystine; however, cystine deficiency is not clinically significant because absorption of short-chain amino acids is not affected.
Normally, cystine and the other dibasic amino acids (ie, ornithine, lysine, arginine) are filtered at the glomerulus and reabsorbed in the proximal convoluted tubule by a high-affinity luminal transmembrane channel. Defects in this channel cause elevated levels of dibasic amino acid secretion in the urine. Whereas ornithine, lysine, and arginine are completely soluble, cystine is relatively insoluble at physiologic urine pH levels of 5-7, with a pKa level of 8.3. At a urine pH level of 7.8 and 8, the respective solubility of cystine is nearly doubled and tripled.
Dent and Senior demonstrated that the solubility of cystine is pH-dependent. [18] The solubility of cystine in urine is approximately 250 mg/L (1 mmol/L) up to a pH level of 7, but solubility increases with a higher pH level by up to 500 mg/L (2 mmol/L) or more above a pH level of 7.5, as depicted in the image below. Measurements in urine have clearly shown that cystine solubility increases linearly with increased ionic strength. Pak and colleagues showed that approximately 70 mg of additional cystine can be dissolved in each liter of solution, with an increase in ionic strength from 0.005-0.3. [19] In addition, at the same ionic strength and pH, cystine solubility varies depending on the particular type of electrolyte present. See the image below.
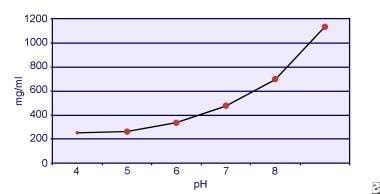
In vitro experiments by Pak and Fuller in 1983 revealed that the highest solubility is accomplished in the presence of calcium chloride, followed by magnesium and sodium chloride. [20] Furthermore, cystine solubility is also affected by urinary macromolecules. The presence of colloid in normal urine has been shown to increase cystine solubility; however, the mechanism of this action is not clear. Because nothing inhibits cystine crystallization, the main determinant is urinary supersaturation. Heterogeneous nucleation of calcium oxalate, brushite, or hydroxyapatite does not occur in individuals with cystinuria.
Risk factors for cystine crystallization include (1) low pH level, (2) reduced ion strength, (3) the presence of cystine crystals, and (4) low levels of urinary macromolecules.
Cystine is a disulfide-linked homodimer of cysteine and has the following structure:
COOH-CHNH2-CH2 –S-S-CH2-CHNH2- COOH Cystine
COOH-CHNH2-CH2 –SH Cysteine
Cystine is absorbed in the small intestine in a manner similar to that of the kidneys. In persons with cystinuria, intestinal absorption of cystine is also impaired to varying degrees. Metabolism of methionine is another source of serum cystine. Two type II membrane glycoproteins domains have been implicated in amino acid transport via the plasma membrane. The first is rBAT, and the second is 4F2HC (the heavy chain of the 4F2 antigen)
Two thirds of persons with cystinuria who form stones make pure cystine calculi, and one third have a mixture of cystine and calcium oxalate calculi. In 2002, Martins et al reported that calcium oxalate precipitation occurs by a salting-out process, ie, the reduction in solubility of a substance due to the addition of another substance to the system, rather than by the process of heterogenous nucleation. [21] Hypocitraturia, hypercalciuria, and hyperuricosuria are also frequently associated with cystinuria. Given their relatively uniform crystalline structure without lamellated cleavage planes, pure cystine calculi are among the hardest on Dretler's stone fragility index.
Cystinuria is an autosomal-recessive disease divided into 3 subtypes: Rosenberg I, II, and III. Cystinuria type I is the most common variant and has been mapped to band 2p16.3. Type I heterozygotes show normal aminoaciduria. Classic cystinuria, types II and III, were thought to be allelic variants, but linkage analyses have revealed type III to be a defect of an uncharacterized gene (SLC7A9) on band 19q13.1. Heterozygotes of types II and III often manifest cystinuria without cystine calculi and may be at increased risk for other types of urolithiasis. Type I heterozygotes are distinguished by normal levels of urinary cystine.
Unlike type I and type II homozygotes, type III homozygotes show an increase in plasma cystine concentration after oral cystine administration. Harris et al reported the complex nature of the genetics of cystinuria by measuring the level of urinary cystine excretion in the parents (obligate heterozygotes) of cystinuria probands and found fully recessive alleles (both parents excreted cystine in the reference range) and dominant alleles (both parents excreted cystine at high levels). [10]
To classify cystinuria clinically, urinary cystine can be measured in each parent of a proband as phenotype I (recessive, urinary cystine level < 100 µmol/g of creatinine), phenotype II (dominant, urinary cystine level >1000 µmol/g of creatinine), and phenotype III (partially dominant, urinary cystine level 100-1000 µmol/g of creatinine). Cystinuria can also be classified based on the age at which symptoms first appear (ie, infantile, juvenile, adolescent).
In healthy individuals, the upper limit for cystine excretion is 20 mg/g of creatinine (< 10 µmol/mmol of creatinine). Homozygotes excrete more than 400 mg/d (1.7 mmol/d), and cystine excretion in homozygous patients is usually 600-1400 mg/d (2.5-5.8 mmol/d). Heterozygotes with type I and III cystinuria excrete less than 200 mg/d (0.8 mmol/d) and do not form stones. Type II heterozygotes excrete up to 200-400 mg/d, but these patients may form stones. The incidence of stone formation increases when urinary cystine concentration exceeds 700 µmol/L (170 mg/L).
Genetics
In recent years, with the advancements in molecular biology, new insights have accumulated regarding the pathophysiology of cystinuria. In 1992, several investigators reported the expression cloning of a 2.3-kilobase renal cDNA (D2H or rBAT) that induced sodium-independent uptake of cystine and the dibasic amino acids in cRNA-injected Xenopus laevis oocytes. The rBAT gene was mapped to chromosome 2 (band 2p21) between D2S119 and D2S288. This gene is now named SLC3A1 in the Genome Database.
Immunohistochemical and in situ hybridization studies revealed that rBAT is expressed in cells of the S3 (pars recta) segment of the proximal tubule and small intestine at the luminal brush-border membrane. In 1995, Gasparini et al reported that mutations in SLC3A1 occurred in patients with type I cystinuria and not in patients with type II or III cystinuria. [22] To date, more than 160 different mutations have been described, including both small and large deletions of DNA base pairs from the gene. [23] One of the most common genetic alterations in SLC3A1 is called M467T, and most mutations tend to be population-specific. The M467T mutation is fairly common in Mediterranean populations. Interestingly, it accounted for 40% of mutations in a Spanish cohort of families and was rare in patients studied in Quebec, Canada.
In 1999, the SLC7A9 (BAT1) gene was isolated. The gene encodes a 487–amino acid protein and was mapped to chromosome 19 (band 19q13) between D19S414 and D19S220. The BAT1 product appears to be a membrane protein with 12 membrane-spanning regions. Mutations in the BAT1 gene probably cause non–type I cystinuria (Rosenberg type II and III). Mutations at the 19q locus are especially common among Libyan Jews, and the risk of urolithiasis in patients who inherit 2 such 19q locus mutations is roughly comparable to that in patients who inherit 2 rBAT mutations.
116 mutations in this gene have been reported. [23] The most common mutation in Libyan Jews resulted in a methionine replacing the valine amino acid residue (V170M) in the protein. In heterozygotes with the V170M mutation, urinary cystine concentrations range from 86-1238 µmol/g of creatinine. Thus, some of the values in V170M heterozygotes are consistent with type III cystinuria and others with type II cystinuria.
An apparent distinguishing feature between type II and type III cystinuria is the lack of intestinal cystine absorption in type II homozygotes. In 2000, Pras suggested a new classification based on molecular analysis. [24] Recently, Dello Strologo et al have proposed a new genetic classification, as follows: [25]
-
Type A, mutation of both alleles of SLC3A1: Heterozygotes show a normal amino acid urinary pattern.
-
Type B, mutation of both alleles of SLC7A9: Heterozygotes usually show an increase of cystine and dibasic amino acid urinary excretion.
-
Type AB, cystinuria caused by 1 mutation in SLC3A1 and 1 mutation in SLC7A9: Mixed-type cystinuria may be caused by the interaction of 2 distinct mutant genes, and the protein encoded by the 19q gene directly interacts with rBAT in the S3 segment of the proximal tubule (see the Table).
Martens et al (2008) recently reported 3 gene-deletion syndromes associated with type A cystinuria: 2p21 deletion syndrome, hypotonia-cystinuria syndrome (HCS), and an atypical form of hypotonia-cystinuria syndrome. Both alleles of SLC3A1 and PREPL are missing in patients with HCS. An additional gene (C2orf34) is deleted in atypical HCS. [26]
Table. Classification of Cystinuria (Open Table in a new window)
Rosenberg et al [27] |
Type I |
Type II |
Type III |
Molecular |
Type I |
Non–Type I |
|
Responsible gene |
SLC3A1 |
SLC7A9 |
|
Band |
2p21 |
19q13.1 |
|
No. of mutations |
>60 |
39 |
|
Most common mutation |
M467 |
V170M |
|
Population affected |
Mediterranean Spanish persons, 40% |
Libyan Jews |
|
Deletion rate |
54% |
25% |
|
Protein |
rBAT |
BAT1 |
|
Amino acid transport system |
|||
Localization in proximal converted tubule |
S3 |
S1, S2 |
|
Transporter characteristic |
High affinity, low capacity |
Low affinity, high capacity |
|
Clinical features |
|||
Homozygotes |
Symptomatic |
approximately 90% symptomatic |
|
Heterozygotes |
Asymptomatic |
approximately 10%-13% symptomatic |
|
Urinary cystine levels |
Normal |
Elevated +++++ |
Elevated + |
Plasma cystine levels after an oral load test |
Same |
Same or slight rise |
Increased |
Intestinal transport |
Absent (no transport of cystine, lysine, or arginine) |
Absent |
Reduced |
Recent evidence suggests that the 4F2HC/4F2LC complex accounts for the Y+L amino acid transport system at the basolateral surface of intestinal and renal proximal tubular cells and that the mutations of the 4F2LC gene (SLC7A7) on band 14q11-13 cause the rare recessive disease called lysine-protein intolerance.
Summary
rBAT, a 90-kd type II glycoprotein, is a high-affinity, sodium-independent transporter for dibasic amino acids in the proximal convoluted renal tubules in rodents.The human rBAT gene has been localized on band 2p21. Interestingly, linkage analysis suggests that this is the same region to which a cystinuric locus, SLC3A1, has been identified.160 different mutations in the SLC3A1 gene and 116 in the SLC7A9 gene have been identified in patients with cystinuria worldwide. [23]
Type III and II cystinuria (non–type I) have been linked to band 19p13.1 (SLC7A9); however, further studies are needed to determine the exact role of the SLC7A9 gene. Approximately 50% of children with 2 SLC3A1 mutations (classic homozygous type I cystinuria) develop at least one stone within the first decade of life.
Etiology
Cystinuria is an autosomal-recessive disease. Two genes have been implicated, both of which code for proteins involved in the transport of neutral and basic amino acids: SLC3A1 codes for rBAT; and SLC7A9 codes for and b0,+AT. [28] The genetic defect impairs intestinal absorption and renal reabsorption of cystine, causing elevated urinary levels of cystine and subsequent crystallization and stone formation.
Epidemiology
Cystine accounts for 1% of adult and 6%-8% of pediatric urinary calculi in the United States. The estimated prevalence of cystinuria is about 1 in 10,000, equating to approximately 33,000 cases annually. [29]
Worldwide, the overall prevalence is 1 person per 7000 population but varies significantly by population. [30] Prevalences of cystinuria are 1 case in 18,000 in Japan, 1 case in 2500 in Israel, 1 case in 2000 in Great Britain, 1 case in 4000 in Australia, 1 case in 1900 in Spain, 1 case in 2500 in Libyan Jews, 1 case in 100,000 in Sweden, 4.5 in 100,000 in Saudi Arabia. [31] In Canada, the Quebec Genetic Network Neonatal Screening Program reported the incidence of persistent cystinuria as 562 cases per million infants, a rate 7 times higher than for clinically manifested cystinuria in the adult population of Quebec. This suggests that many cystinuric individuals do not form stones.
Men are more severely affected. An annual incidence of 0.42 stone episodes for in males with cystinuria and 0.21 in females with the disease has been reported. [25] Although stones may present at any age, stone presentation most commonly occurs within the first two decades of life, with approximately 50% of cystinuric patients developing their first stone in the first decade of life and 25% to 40% during their teenage years. [29]
Prognosis
No curative treatment of cystinuria exists, and patients will have a life-long risk of stone formation, repeated surgery, and impaired kidney function and quality of life. One study reported 1.22 stone episodes per year. Recurrence rates after surgical intervention approach 45% at 3 months without medical management. The recurrence rate with medical management improves to approximately 25% at 3 years after surgery but is still inferior to rates for other types of calculi. The probability of a recurrence-free survival at 1- and 5-year follow-up is 0.73 and 0.27, respectively. [32] Barbey et al reported one new stone formation per patient per year and an average of one surgical procedure every 3 years, with 7 surgical procedures for nephrolithiasis by middle age. [3]
Urinary calculi are generally the only manifestation of cystinuria, although 10% of cases are complicated by hypertension, and one study found a weak association with short stature. Patients with cystinuria who form stones are at higher risk for anatomical renal loss (nephrectomy) than those who form calcium oxalate stones. The risk of impairment in kidney function is high; up to 70% of patients may be affected, depending on the length of follow-up and medical therapy. However, according to Lindell et al, end-stage renal disease occurs in less than 5% of patients with cystinuria. [33] Other complications include chronic pyelonephritis, mental illness, and intellectual disability. [34]
Patient Education
Cystinuria is an inherited metabolic disorder; therefore, patient education is extremely important. The children of parents who both have cystinuria have a 100% chance of becoming cystinuric. If one parent is fully cystinuric and the other is a carrier, the chance of each child becoming fully cystinuric is 50%. If both parents are carriers, the chance of each child becoming cystinuric is 25%. If one parent is cystinuric and the other is neither cystinuric nor a carrier, the chance of each child becoming cystinuric is nil.
To help prevent recurrence, counsel and educate patients in whom recurrence was caused by medication noncompliance regarding the importance of proper diet and the necessity of medication compliance. [35]
For patient education information, see What to Know About Cystinuria. Further information about cystinuria is available on the following Web sites:
-
Cystine solubility in urine.
-
Electron microscopic picture showing cystine crystals.
-
Plain radiograph of the abdomen showing cystine staghorn stones.
-
Faintly opaque (ground-glass appearance) left lower ureteric stone.
-
Intravenous urogram showing left ureterohydronephrosis.
-
Renal sonogram demonstrating renal calculi in the lower pole.
-
Treatment algorithm for cystinuria.
Tables
Rosenberg et al [27] |
Type I |
Type II |
Type III |
Molecular |
Type I |
Non–Type I |
|
Responsible gene |
SLC3A1 |
SLC7A9 |
|
Band |
2p21 |
19q13.1 |
|
No. of mutations |
>60 |
39 |
|
Most common mutation |
M467 |
V170M |
|
Population affected |
Mediterranean Spanish persons, 40% |
Libyan Jews |
|
Deletion rate |
54% |
25% |
|
Protein |
rBAT |
BAT1 |
|
Amino acid transport system |
|||
Localization in proximal converted tubule |
S3 |
S1, S2 |
|
Transporter characteristic |
High affinity, low capacity |
Low affinity, high capacity |
|
Clinical features |
|||
Homozygotes |
Symptomatic |
approximately 90% symptomatic |
|
Heterozygotes |
Asymptomatic |
approximately 10%-13% symptomatic |
|
Urinary cystine levels |
Normal |
Elevated +++++ |
Elevated + |
Plasma cystine levels after an oral load test |
Same |
Same or slight rise |
Increased |
Intestinal transport |
Absent (no transport of cystine, lysine, or arginine) |
Absent |
Reduced |





