Background
The complement system is part of the innate immune system. The complement system plays an important part in defense against pyogenic organisms. It promotes the inflammatory response, eliminates pathogens, and enhances the immune response. Deficiencies in the complement cascade can lead to overwhelming infection and sepsis.
In addition to playing an important role in host defense against infection, the complement system is a mediator in both the pathogenesis and prevention of immune complex diseases, such as systemic lupus erythematosus (SLE). These findings underscore the duality of the complement system. It has a protective effect when functioning in moderation against pathogens; at the same time, the inflammation promoted by complement activation can result in cellular damage when not kept in check.
Complement deficiencies are said to comprise between 1 and 10% of all primary immunodeficiencies. [1] The genetic deficiency of early components of the classical pathway (C1q, C1r/s, C2, C4) tend to be linked with autoimmune diseases [2] , whereas C5 to C9 may have enhanced susceptibility to meningococcal disease. Some new clinical entities are linked with partial complement defects. C2 deficiency is associated with an increased risk of infections with encapsulated bacteria, such as Haemophilus influenzae. [3]
Cases of complement deficiency have helped defined the role of complement in host defense. [4] A registry of complement deficiencies has been established as a means to promote joint projects on treatment and prevention of diseases associated with defective complement function. Knowledge about the complement system is expanding. New studies point to the complex interplay between the complement cascade and adaptive immune response, and complement is also being studied in association with ischemic injury as a target of therapy. Although the complement system is part of the body's innate, relatively nonspecific defense against pathogens, its role is hardly primitive or easily understood. This article outlines some of the disease states associated with complement deficiencies and their clinical implications. [5]
Genes that encode the proteins of complement components or their isotypes are distributed throughout different chromosomes, with 19 genes comprising 3 significant complement gene clusters in the human genome. [6] Genetic deficiency of C1q, C1r/s, C2, C4, and C3 is associated with autoimmune diseases, whereas deficiency of C5, C6, C7, C8, C9 increase susceptibility to infections.
Pathophysiology
The complement cascade consists of 3 separate pathways that converge in a final common pathway. The pathways include the classical pathway (C1qrs, C2, C4), the alternative pathway (C3, factor B, properdin), and the lectin pathway (mannan-binding lectin [MBL]). The classical pathway is triggered by interaction of the Fc portion of an antibody (immunoglobulin [Ig] M, IgG1, IgG2, IgG3) or C-reactive protein with C1q. The alternative pathway is activated in an antibody-independent manner. Lectins activate the lectin pathway in a manner similar to the antibody interaction with complement in the classical pathway. These 3 pathways converge at the component C3. Although each branch is triggered differently, the common goal is to deposit clusters of C3b on a target. This deposition provides for the assembly of the membrane attack complex (MAC), components C5b-9. The MAC exerts powerful killing activity by creating perforations in cellular membranes. See the image below.
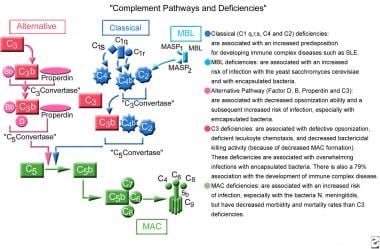 Complement pathways and deficiencies.
Complement pathways and deficiencies.
Deficiencies in complement predispose patients to infection via 2 mechanisms: (1) ineffective opsonization and (2) defects in lytic activity (defects in MAC). Specific complement deficiencies are also associated with an increased risk of developing autoimmune disease, such as SLE.
An intricate system regulates complement activity. The important components of this system are various cell membrane–associated proteins such as complement receptor 1 (CR1), complement receptor 2 (CR2), and decay accelerating factor (DAF). A North African study of molecular basis of complement factor I deficiency in atypical hemolytic and uremic syndrome patients suggested that the Ile357Met mutation may be a founding effect. [7]
In addition to these cell surface–associated proteins, other plasma proteins regulate specific steps of the classic or alternative pathway; for example, the proteins factor H and factor I inhibit the formation of the enzyme C3 convertase of the alternative pathway. Similarly, the enzyme C1q esterase acts as an inhibitor of the classic pathway serine proteases C1r and C1s. Deficiency of any of these regulatory proteins results in a state of overactivation of the complement system, with damaging inflammatory effects. [8] Two clinical manifestations of such deficiencies are paroxysmal nocturnal hemoglobinuria and hereditary angioedema, both of which are discussed in other Medscape Reference articles (see Paroxysmal Nocturnal Hemoglobinuria and Angioedema). A sampling of thirteen hereditary angioedema patients, mainly female and younger than 50 years, with deficiency or normal C1 inhibitor who experienced COVID-19 had neither severe acute angioedema nor severe COVID-19. [9]
Epidemiology
Frequency
Complement deficiencies are relatively rare worldwide; estimates of prevalence are based on results from screening high-risk populations. Retrospective studies of persons with frequent meningococcal infections report varying prevalence based on geographic location. In populations with recurrent meningococcal infection, the prevalence rate is as high as 30%. Individuals with C1q deficiency have a 93% chance of developing SLE. Similarly, C1rs deficiency has a 57% association with SLE and C4 deficiency has a 75% association with SLE.
Mortality/Morbidity
Individuals with complement deficiencies that hinder opsonization present with frequent recurrent infections and a high rate of morbidity and mortality. Deficiency of C3, the major opsonin, results in recurrent pyogenic infections, particularly with encapsulated bacteria.
Deficiencies of early classical pathway components (C1, C4, C2) do not usually predispose individuals to severe infections but are associated with autoimmune disorders, especially SLE.
Patients with a defect in formation of the MAC have a lesser degree of morbidity and mortality than, for example, patients with a defect in C3; the deficiency in the lytic component of the complement cascade is thought to have some protective effect against the generation of full-blown sepsis. These patients are at high risk for recurrent infection with Neisseria gonorrhoeae or Neisseria meningitidis. Severe pyogenic infections and sepsis occur in children and neonates who have a deficiency of a MAC component.
Demographics
While no definitive racial patterns of association have been established for the majority of complement deficiencies, ethnic predispositions have been described for certain complement deficiencies. For example, deficiencies in properdin and C2 have been associated with the white race, C6 deficiencies have been shown to have a possible predisposition in African populations, and deficiencies in C8 and C9 have been associated with an Asian racial background. More specifically, 2 functionally distinct C8 deficiency states have been described: C8 alpha-gamma deficiency seen mostly in persons of Afro-Caribbean, Hispanic, and Japanese descent; and C8beta, mainly evident in persons of Caucasian descent. [10] However, for most of these deficiencies, the absolute number of patients studied has been quite small.
Most complement deficiencies affect both sexes equally.
The majority of complement deficiencies are inherited in an autosomal recessive pattern (although MBL deficiency has been described as having both an autosomal dominant and recessive pattern). An exception to the autosomal pattern of inheritance is properdin deficiency, which is an X-linked trait.
Individuals with complement deficiencies that hinder opsonization often present at an early age (months to a few years) because of increased susceptibility to overwhelming infection.
Patients with deficiencies in formation of the MAC tend to present when slightly older (late-teenage years).
Complement deficiencies associated with immune complex diseases, such as SLE, do not show a clear pattern of age at first presentation.
-
Complement pathways and deficiencies.




