Practice Essentials
Acute tubular necrosis (ATN) is the most common cause of acute kidney injury (AKI) in the renal category (that is, AKI in which the pathology lies within the kidney itself). The term ATN is actually a misnomer, as there is minimal cell necrosis and the damage is not limited to tubules. [1] See the ATN image below.
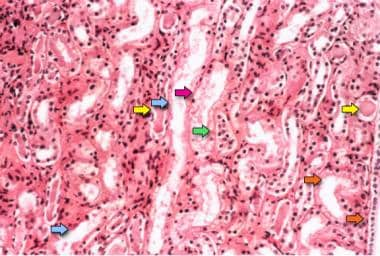 Acute tubular necrosis. Photomicrograph of a kidney biopsy specimen shows renal medulla, which is composed mainly of renal tubules. Features suggesting acute tubular necrosis are the patchy or diffuse denudation of the renal tubular cells with loss of brush border (blue arrows); flattening of the renal tubular cells due to tubular dilation (orange arrows); intratubular cast formation (yellow arrows); and sloughing of cells, which is responsible for the formation of granular casts (red arrow). Finally, intratubular obstruction due to the denuded epithelium and cellular debris is evident (green arrow); note that the denuded tubular epithelial cells clump together because of rearrangement of intercellular adhesion molecules.
Acute tubular necrosis. Photomicrograph of a kidney biopsy specimen shows renal medulla, which is composed mainly of renal tubules. Features suggesting acute tubular necrosis are the patchy or diffuse denudation of the renal tubular cells with loss of brush border (blue arrows); flattening of the renal tubular cells due to tubular dilation (orange arrows); intratubular cast formation (yellow arrows); and sloughing of cells, which is responsible for the formation of granular casts (red arrow). Finally, intratubular obstruction due to the denuded epithelium and cellular debris is evident (green arrow); note that the denuded tubular epithelial cells clump together because of rearrangement of intercellular adhesion molecules.
ATN follows a well-defined four-part sequence of initiation, extension, maintenance, and recovery, as follows:
-
The initiation phase is characterized by an acute decrease in glomerular filtration rate (GFR), with a corresponding sudden increase in serum creatinine (Cr) and blood urea nitrogen (BUN) concentrations.
-
The extension phase is characterized by ongoing hypoxia leading to sustained tissue ischemia, which triggers an inflammatory response. Given its limited blood supply, the corticomedullary junction of the kidney is most susceptible to a renal insult. A continuous release of cytokines and chemokines from impaired corticomedullary junction enhances the inflammatory cascade—and this ongoing activation of the inflammatory cascade results in the drop of GFR.
-
The maintenance phase is characterized by a sustained reduction in GFR that persists for a variable length of time, most commonly 1-2 weeks. Because of the low GFR, the Cr and BUN levels continue to rise. Cellular repair from injury starts in this phase.
-
The recovery phase, in which tubular function recovers, is characterized by an increase in urine volume (if oliguria was present during the maintenance phase) and by a gradual decrease in BUN and serum Cr to their preinjury levels.
See Pathophysiology for more detail.
The tubule cell damage and cell death that characterize ATN usually result from an acute ischemic or toxic event. Nephrotoxic mechanisms of ATN include direct drug toxicity, intrarenal vasoconstriction, and intratubular obstruction (see Pathophysiology and Etiology). Whether the cause is ischemia or nephrotoxicity, most of the pathophysiologic features of ATN are the same. [2]
The history, physical examination, and laboratory and imaging findings, especially renal ultrasonogram and urinalysis, are particularly helpful in identifying the cause of ATN (see Presentation and Workup).
Therapeutic mainstays are prevention, avoidance of further kidney damage, treatment of underlying conditions, and aggressive treatment of complications (see Treatment and Medication).
Go to Pediatric Acute Tubular Necrosis for complete information on this topic. For patient education information, see Acute Kidney Failure.
Pathophysiology
ATN follows a well-defined four-part sequence of initiation, extension, maintenance, and recovery (see below). [3, 4] The tubule cell damage and cell death that characterize ATN usually result from an acute ischemic or toxic event. [5] The ischemic and the nephrotoxic forms of ATN share most of the same pathophysiologic features.
Initiation
Ischemic ATN is often described as a continuum of prerenal azotemia. Indeed, the causes of the two conditions are the same. Ischemic ATN results when hypoperfusion overwhelms the kidney’s autoregulatory defenses. Under these conditions, hypoperfusion initiates cell injury that often, but not always, leads to cell death.
Extension
Persistent hypoperfusion injures tubular cells in the straight portion of the proximal tubules and the thick ascending limb of the loop of Henle, especially as it dips into the relatively hypoxic medulla. The tubular epithelial cells of the S3 segment of the proximal tubule in the outer stripe of the medulla, at the corticomedullary junction, are most susceptible to ischemic injury as these cells live in a relatively hypoxic environment.
Continued hypoperfusion can also cause vascular endothelial injury and prolong the extension phase with compromised blood flow to injured tissues and inability to regulate the local vascular tone. As cells in segments S1 and S2 possess high rates of endocytosis, the injury from exogenous toxins is more commonly seen in S1 and S2 segments. The reduction in GFR that occurs from ischemic injury is a result not only of reduced filtration due to hypoperfusion but also of casts and debris obstructing the tubule lumen. The infiltrate thus leaks back through the damaged epithelium, resulting in ineffective filtration.
The earliest changes in the proximal tubular cells are apical blebs and loss of the brush border membrane, followed by a loss of polarity and integrity of the tight junctions. This loss of epithelial cell barrier can result in the back-leak of filtrate mentioned above.
Another change is the relocation of Na+/K+-ATPase pumps and integrins to the apical membrane. Cell death occurs by both necrosis and apoptosis. Sloughing of live and dead cells occurs, leading to cast formation and obstruction of the tubular lumen (see the image below). Activation of the renal immune system—with damage to tubular cells stimulating local secretion of proinflammatory cytokines—in turn induces further necrosis. [6]
Acute tubular necrosis. Photomicrograph of a kidney biopsy specimen shows renal medulla, which is composed mainly of renal tubules. Features suggesting acute tubular necrosis are the patchy or diffuse denudation of the renal tubular cells with loss of brush border (blue arrows); flattening of the renal tubular cells due to tubular dilation (orange arrows); intratubular cast formation (yellow arrows); and sloughing of cells, which is responsible for the formation of granular casts (red arrow). Finally, intratubular obstruction due to the denuded epithelium and cellular debris is evident (green arrow); note that the denuded tubular epithelial cells clump together because of rearrangement of intercellular adhesion molecules.
In addition, ischemia leads to decreased production of vasodilators (ie, nitric oxide, prostacyclin [prostaglandin I2, or PGI2]) by the tubular epithelial cells, worsening vasoconstriction and hypoperfusion. On a cellular level, ischemia causes depletion of adenosine triphosphate (ATP), an increase in cytosolic calcium, formation of free radicals, metabolism of membrane phospholipids, and abnormalities in cell volume regulation.
The decrease or depletion of ATP leads to many problems with cellular function, not the least of which is active membrane transport. Ineffective active membrane transport disrupts cell volume and electrolyte regulation, causing cell swelling and intracellular accumulation of sodium and calcium. Typically, phospholipid metabolism is altered, and membrane lipids undergo peroxidation. In addition, free radical formation is increased, producing toxic effects. Damage inflicted by free radicals is most severe during reperfusion.
Maintenance
The maintenance phase of ATN is characterized by a stabilization of GFR, typically lasting 1-2 weeks. Complications (eg, uremic and others; see Complications) typically develop during this phase.
The mechanisms of injury described above may contribute to continued nephron dysfunction, but tubuloglomerular feedback also plays a role. Tubuloglomerular feedback in this setting leads to constriction of afferent arterioles by the macula densa cells, which detect an increased salt load in the distal tubules. The resulting decreased perfusion of the nephrons can perpetuate the kidney injury.
Recovery
The recovery phase of ATN is characterized by regeneration of tubular epithelial cells. [7] During recovery, abnormal diuresis sometimes occurs, causing salt and water loss and volume depletion. The mechanism of the diuresis is not entirely understood. Still, it may, in part, be due to the delayed recovery of tubular cell function in the setting of increased glomerular filtration. In addition, continued use of diuretics (often administered during initiation and maintenance phases) may also add to the problem.
ATN is reversible in some patients but not in all patients. The underlying mechanism is not very clear, especially the molecular switch. Upon severe or persistent kidney injury, the repair is incomplete or maladaptive. Maladaptive repair could result in permanent injury or kidney fibrosis. Various cell types and multiple injury pathways are involved in maladaptive repair, but the critical component is inflammation. Renal tubular cells could release cellular contents and cytokines to surrounding tissue and trigger an inflammatory response. Thus, the sustained necroinflammatory process could cause severe organ dysfunction and tissue injury in the kidney [8] .
Further, at molecular levels, regulated cell death through tubular cell apoptosis, necrosis, and cell loss drive AKI development upon a renal insult. Apoptosis, necroptosis, ferroptosis, MPT-RN (mitochondrial permeability transition regulated necrosis), and possible pyroptosis are potential molecular mechanisms of AKI induced by ischemic-reperfusion injury, toxin, cytokine storm, and crystals [9] .
The driving force of ATN is primarily tubular injury and hypoperfusion in peritubular capillaries. The combination of tubular injury and hypoperfusion could cause permanent nephron loss in those patients who cannot recover from ATN, eventually tissue fibrosis and CKD after acute injury.
Normotensive ischemic acute tubular necrosis
This condition develops in patients without an overt severe hypotensive episode. These patients have low to normal blood pressure but still have severe ATN. The most common reason for this condition is renal susceptibility to lower blood pressure because of impairment of autoregulatory function of the kidney. Normally, the afferent arteriole dilates (via prostaglandins) and efferent arteriole constricts (via angiotensin-II) to maintain the glomerular capillary pressure. Factors that impair this autoregulatory mechanism include the following [10] :
-
Advanced age
-
Atherosclerosis, hypertension, and chronic kidney disease
-
Malignant hypertension
-
Medications impairing the autoregulatory mechanism (e.g., nonsteroidal anti-inflammatory drugs [NSAIDs])
-
Afferent glomerular vasoconstriction (e.g., from sepsis, hypercalcemia, hepatorenal syndrome, cyclosporine/tacrolimus)
-
Drugs blocking efferent arteriolar vasoconstriction - Angiotensin-converting enzyme (ACE) inhibitors/angiotensin receptor blockers (ARBs)
Endotoxemia (sepsis)-related ATN
Sepsis is a recognized cause of ATN. Development of AKI in the setting of sepsis will increase in-hospital mortality by five to six-fold [11, 12] and increase the risk of progression to CKD in survivors [13] . Sepsis-induced ATN is hypothesized from a reduction in renal blood flow caused by sepsis-related hypotension. However, this has been challenged by several animal and human studies. These studies have indicated that, in fact, renal blood flow may increase in that setting due to a mechanism that leads to afferent arteriolar vasodilatation. [14]
Other suspected contributors to ATN in sepsis include the following: [15, 16, 17, 18]
-
Intra-renal vasoconstriction and redistribution of renal blood flow to the cortex
-
Activation of vasoactive intrarenal hormones (endothelin, renin-angiotensin- aldosterone)
-
Nitric oxide synthase induction and release of several cytokines
-
Local cytokine release that sustains an inflammatory response
Etiology
ATN is generally caused by an acute event, either ischemic or toxic. ATN is caused by sepsis in approximately 20% of intensive care unit (ICU) patients. Prerenal azotemia, obstruction, glomerulonephritis, vasculitis, acute interstitial nephritis, acute on chronic injury (in patients with chronic kidney disease [CKD]), and atheroembolic injury account for most of the remainder. [19, 20]
Causes of ischemic acute tubular necrosis
Ischemic ATN may be considered part of the spectrum of prerenal azotemia; indeed, ischemic ATN and prerenal azotemia have the same causes and risk factors. Specifically, these include the following:
-
Hypovolemic states: Hemorrhage, volume depletion from gastrointestinal (GI) or renal losses, burns, fluid sequestration
-
Low cardiac output states: Heart failure and other diseases of myocardium, valvulopathy, arrhythmia, pericardial diseases, tamponade
-
Systemic vasodilation: Sepsis, anaphylaxis
-
Surgical procedures in which renal blood flow is compromised (eg, those that involve clamping of the renal artery, such as coronary artery bypass grafting [CABG], aortic dissection repair, renal cell carcinoma resection)
Causes of ischemic ATN vary depending on the studied population. In developed countries, comorbidities, older age, and severity of illness during ICU admission are more likely to result in AKI, as reported in the Acute Kidney Injury–Epidemiologic Prospective Investigation (AKI-EPI) [21] and Assessment, Serial Evaluation, and Subsequent Sequelae of Acute Kidney Injury (ASSESS-AKI) trials. [22, 23] These comorbidities include cancer, hypertension, chronic heart failure, cirrhosis, AIDS, chronic obstructive pulmonary disease, and diabetes mellitus. In low-income countries or rural areas, AKI occurs more commonly in younger patients and in association with conditions related to volume depletion and complications of pregnancy. [24] However, there is no detailed information on ATN in those settings.
Causes of nephrotoxic acute tubular necrosis
The kidney is a particularly vulnerable target for toxins, both exogenous and endogenous. Not only does it have a rich blood supply, receiving 25% of cardiac output, but it also helps in the excretion of these toxins by glomerular filtration and tubular secretion.
Exogenous nephrotoxins that cause ATN
Aminoglycoside-related toxicity occurs in 10-30% of patients receiving aminoglycosides, even when blood levels are in apparently therapeutic ranges. Risk factors for ATN in these patients include the following:
-
Preexisting liver or kidney disease
-
Concomitant use of other nephrotoxins (eg, amphotericin B, radiocontrast media, cisplatin)
-
Shock
-
Advanced age
-
Female sex
-
Higher aminoglycoside level 1 hour after dose (a high trough level has not been shown to be an independent risk factor)
Amphotericin B nephrotoxicity risk factors include the following:
-
Male sex
-
High maximum daily dose (nephrotoxicity is more likely to occur if > 3 g is administered)
-
Longer duration of therapy
-
Hospitalization in the critical care unit at the initiation of therapy
-
Concomitant use of cyclosporine
Radiographic contrast media can cause contrast-induced nephropathy (CIN). This commonly occurs in patients with several risk factors, including preexisting CKD, underlying diabetic nephropathy, chronic heart failure (CHF), or high or repetitive doses of contrast media, as well as volume depletion and concomitant use of diuretics, angiotensin-converting enzyme inhibitors (ACEi), or angiotensin II receptor blockers (ARBs). Prevention is paramount, so patients at risk of CIN should have a careful evaluation of volume status and receive volume expansion with 0.9% sodium chloride or crystalloids with less chloride content before the procedure. [25]
Other exogenous nephrotoxins that can cause ATN include the following:
-
Cyclosporine and tacrolimus (calcineurin inhibitors)
-
Cisplatin
-
Ifosfamide
-
Foscarnet
-
Pentamidine, which is used to treat Pneumocystis jiroveci infection in immunocompromised individuals (risk factors for nephrotoxicity include volume depletion and concomitant use of other nephrotoxic antibiotic agents, such as aminoglycosides, which is common practice in the immunosuppressed)
-
Sulfa drugs
-
Acyclovir and indinavir
-
Tenofovir
-
Mammalian (mechanistic) target of rapamycin (mTOR) inhibitors (eg, everolimus, temsirolimus) [26]
Endogenous nephrotoxins that cause ATN
In myoglobinuria, rhabdomyolysis is the most common cause of heme pigment–associated AKI and can result from traumatic or nontraumatic injuries. Most cases of rhabdomyolysis are nontraumatic, such as those related to alcohol abuse or drug-induced muscle toxicity (eg, statins alone or in combination with fibrates).
In hemoglobinuria, AKI is a rare complication of hemolysis and hemoglobinuria. It is often associated with transfusion reactions (in contrast to myoglobin, hemoglobin has no apparent direct tubular toxicity, and AKI in this setting is probably related to hypotension and decreased renal perfusion). [26] A few reports suggest intratubular casts as a culprit of ATN in patients with significant hemolysis. [27]
Acute crystal-induced nephropathy occurs when crystals are generated endogenously due to high cellular turnover (ie, uric acid), as observed in certain malignancies or the treatment of malignancies. However, this condition is also associated with ingestion of certain toxic substances (eg, ethylene glycol) or nontoxic substances (eg, vitamin C). Choudhry et al reported an AKI case caused by ingesting excessive amounts of calcium-containing antacids. [28]
In multiple myeloma, renal impairment results from the accumulation and precipitation of light chains, which form casts in the distal tubules that cause renal obstruction. In addition, myeloma light chains have a direct toxic effect on proximal renal tubules. [29]
COVID-19
AKI is one of the most common complications of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (COVID-19) in hospitalized patients, occurring in about 25% to 50% of cases. [30, 31, 32, 33] In one study, only a third of all patients with AKI were discharged with recovery of kidney function.{ref64 Among all patients with AKI, 19-33.8% required renal replacement therapy. [34, 35] Preexisting medical conditions are associated with higher AKI incidence in COVID patients. Among patients with CKD, the incidence of AKI was close to 80%. Similar to the causes of AKI in other patient populations, the common risk factors for COVID-related AKI are old age, diabetes mellitus, hypertension, CKD, chronic heart failure, liver disease, male gender, obesity, and severe hypoxia on presentation. [36, 37, 38, 39, 40]
COVID-19 infection can cause proteinuria in 39-81% of AKI patients. Hematuria and leukocyturia are seen in 26-84% and 60% of COVID-AKI patients, respectively. Some patients develop nephrotic range proteinuria. The causes of AKI are commonly pre-renal, ischemic, and toxic ATN from hemodynamic instability, rhabdomyolysis, contrast exposure, or drugs. [41, 42] The major cause of AKI in COVID-19 is ATN. The pathological changes in COVID-AKI are mainly acute tubular injury (ATI); pigmented casts from rhabdomyolysis; different types of vascular injury, including frequent red blood cell aggregation occluding renal capillaries, endothelial swelling and foaming degeneration; and segmental fibrin thrombi from the hypercoagulable state. Hypercoagulation is frequently seen in COVID-19 infection. [43, 44]
AKI in patients with COVID-19 is also associated with significantly higher mortality.
Epidemiology
The landmark PICARD (Program to Improve Care in Acute Renal Disease) study was an observational study of a cohort of 618 patients with AKI in the intensive care units of 5 academic centers in the United States. Ischemic ATN was the presumed etiology for 50% of all patients with renal failure, an additional ~12% due to unresolved pre-renal factors, and ~25% from nephrotoxic ATN. [45] These data were similar to those from the Madrid Acute Renal Failure Study Group, which assessed 748 cases of acute injury from that region of Spain and estimated that the incidence of ATN was 88 cases per million population. [46]
Prognosis
For patients with ATN, the in-hospital survival rate is approximately 50%, with about 30% surviving for one year. Critically ill patients with severe AKI have higher mortality in the first two months and less during long-term follow-up. [47]
Factors associated with an increased mortality rate in patients with ATN include the following:
-
Poor nutritional status
-
Male sex
-
Oliguria
-
Need for mechanical ventilation
-
Acute myocardial infarction
-
Stroke
-
Seizures
The mortality rate in patients with ATN is probably related more to the severity of the underlying disease than to ATN itself. For example, the mortality rate in patients with ATN after sepsis or severe trauma is much higher (about 60%) than in patients with nephrotoxin-related ATN (about 30%). The mortality rate is as high as 60-70% with patients in a surgical setting. If multiorgan failure is present, especially severe hypotension or acute respiratory distress syndrome, the mortality rate ranges from 50% to 80%.
Patients with oliguric ATN have a worse prognosis than patients with non-oliguric ATN. This probably is related to more severe necrosis and more significant disturbances in electrolyte balance. In addition, a rapid increase in serum Cr to a high level (ie, > 3 mg/dL) indicates a poorer prognosis. Again, this probably reflects more serious underlying injury.
Of the survivors of ATN, approximately 50% have some impairment of kidney function. About 5% continue to undergo a decline in kidney function. About 5% never recover kidney function and require dialysis.
A review of United States Renal Data System (USRDS) data (n = 1,070,490) for 2001 through 2010 found that although the incidence of end-stage renal disease (ESRD) attributed to ATN increased during that period, the prospects for renal recovery and survival also increased. Recovery of kidney function was more likely in patients with ATN than in matched controls (patients with ESRD but not ATN; cumulative incidence 23% vs 2% at 12 weeks, 34% vs 4% at 1 year), as was death (cumulative incidence 38% vs 27% at 1 year). Hazard ratios for death declined in stepwise fashion to 0.83 in 2009-2010. [48]
AKI in hospital inpatients has long-term implications after discharge. A meta-analysis of 13 cohort studies showed that patients with AKI had a higher risk for developing CKD (hazard ratio [HR] 8.8, 95% confidence index [CI] 3.1-25.5), ESRD (HR 3.1, 95%CI 1.9-5.0) and mortality (HR 2.0, 95% CI 1.3 - 3.1). [49] AKI was also independently associated with cardiovascular disease and CHF risk. Patients with AKI whose kidney function does not return to 25% of baseline have a higher risk of mortality and adverse renal outcomes. [50]
The United States Renal Data System (USRDS) annual report demonstrated that AKI-related hospital admission rates in Medicare recipients decreased for the first time in 2020, except in Blacks. [51] However, the percentage of hospitalized patients requiring dialysis for AKI increased, to 4.1% overall (ranging from 6.2% in those aged 66-69 years to1.8% in those aged ≥85 years).
Rates of death or entrance into hospice care in patients with AKI ranged from about 17.5% for those not requiring dialysis to 41.8% for those requiring dialysis and 74.1% for those with COVID-19. After hospital discharge, an additional 13.8% died and 10.7% required re-admission. Of beneficiaries with pre-existing CKD, 17.5% progressed to advanced CKD, and 42.9% died two years after index hospitalization for AKI. Among patients with AKI requiring hemodialysis, only 29.7% recovered kidney function, 18.8% remained on dialysis, 35.7% progressed to ESRD, and 15.8% died by three months. Only 32.2% recovered kidney function by six months; 2.1% remained on hemodialysis, 48.8% progressed to ESRD, and 16.9% died. [51]
A retrospective study by Wald et al found that up to 25% of critically ill adult patients with AKI required dialysis. [52] Pre-existing CKD was associated with a higher rate of dialysis requirement. Worsening kidney function developed in the first months of follow-up and increased substantially three years after a severe AKI episode. The RENAL trial reported a similar long-term survival rate of 38%; most deaths occurred in the first three months.
Co-morbidities are the main factors affecting life expectancy after severe AKI attacks. [53, 54] In the ASSESS-AKI trial and other studies, about 5.7% to 9% of patients had kidney disease progression. Higher post-AKI urine albumin Cr ratio (ACR) was associated with the progression of kidney disease at median follow-up of 4.7 years (hazard ratio 1.53, 95% CI 1.45-1.62). [22, 55]
-
Acute tubular necrosis. Pigmented, muddy brown, granular casts are visible in the urine sediment of a patient with acute tubular necrosis (400x magnification).
-
Acute tubular necrosis. Photomicrograph of a kidney biopsy specimen shows renal medulla, which is composed mainly of renal tubules. Features suggesting acute tubular necrosis are the patchy or diffuse denudation of the renal tubular cells with loss of brush border (blue arrows); flattening of the renal tubular cells due to tubular dilation (orange arrows); intratubular cast formation (yellow arrows); and sloughing of cells, which is responsible for the formation of granular casts (red arrow). Finally, intratubular obstruction due to the denuded epithelium and cellular debris is evident (green arrow); note that the denuded tubular epithelial cells clump together because of rearrangement of intercellular adhesion molecules.
Tables
| Stage | Serum Creatinine | Urine output |
|---|---|---|
| 1 | 1.5-1.9 times baseline | < 0.5 ml/kg/hr for 6 - 12 hours |
| OR | ||
| ≥ 0.3 mg/dl (≥ 25.5 μmol/l) increase | ||
| 2 | 2.0-2.9 times baseline | < 0.5 ml/kg/hr for ≥ 12 hours |
| 3 | 3.0 times baseline | < 0.3 ml/kg/hr for ≥ 24 hours |
| OR | OR | |
| Increase in serum creatinine to ≥ 4 mg/dl (≥ 353.6 μmol/l) | Anuria for ≥ 12 hours | |
| OR | ||
| Initiation of renal replacement therapy | ||
| OR, inpatients < 18 years, decrease in eGFR to < 35 ml/min per 1.73 m2 |
Finding |
Prerenal Azotemia |
ATN and/or Intrinsic Renal Disease |
Urine osmolarity (mOsm/kg) |
>500 |
< 350 |
Urine sodium (mmol/d) |
< 20 |
>40 |
Fractional excretion of sodium (FENa) (%) |
< 1 |
>2 |
Fractional excretion of urea (%) |
< 35 |
>50 |
Urine sediment |
Bland and/or nonspecific |
May show muddy brown granular casts |