Practice Essentials
The rare variant of congenital adrenal hyperplasia (CAH) known as 17-hydroxylase deficiency was first described in the 1960s in patients with sexual infantilism and hypertension. It has also been described to present in the setting of male pseudohermaphroditism. [1, 2, 3] Patients with 17-hydroxylase deficiency have alterations in their CYP17 gene, which encodes the P450C17 enzyme. [4, 5, 6, 7] This enzyme plays a central role in steroidogenesis. See the image below. Steroidogenesis is essential for the production of cortisol and sex steroids. Thus, patients with 17-hydroxylase deficiency have reduced secretion of cortisol, androgen, and estrogen, with adrenal and gonadal steroidogenesis impairment. Although patients with 17-hydroxylase deficiency have decreased cortisol production, they do not have signs or symptoms of adrenal insufficiency due to elevations of corticosterone and glucocorticoids.
See the image below.
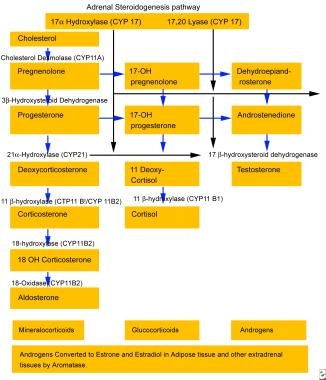
CAH due to 17-hydroxylase deficiency is associated with hypertension and an excess of deoxycorticosterone (DOC), which is the second most common naturally occurring mineralocorticoid after aldosterone. DOC excess typically is associated with hypertension, hypokalemia, and renin and aldosterone suppression. Among the conditions associated with DOC excess are Cushing syndrome (particularly the ectopic adrenocorticotropic hormone [ACTH] variants and in the setting of adrenocortical carcinomas), adrenal tumors, CAH due to 11-hydroxylase deficiency, and primary cortisol resistance. [8, 9]
Pathophysiology
In the zona fasciculata, the typical end-product of the steroid biosynthetic pathway is cortisol, as shown in the image below, which regulates pituitary ACTH production through negative feedback inhibition. Loss of 17-hydroxylase activity in the adrenal gland blocks the synthesis of cortisol and results in an increase in ACTH production. See the image below.
Aldosterone is the main mineralocorticoid produced by the adrenal zona glomerulosa, and its production is regulated by the renin-angiotensin system. A 17-hydroxy pathway similar to the active pathway in the zona glomerulosa occurs in the zona fasciculata; however, the final product is corticosterone rather than aldosterone. In the glomerulosa, but not in the fasciculata, corticosterone is hydroxylated and oxidized at the 18 position to produce aldosterone. The adrenal fasciculata's production of corticosterone, a weak glucocorticoid, and DOC, a potent mineralocorticoid, is minimal and relatively unimportant in healthy, normal individuals, but it is important in patients with 17-hydroxylase deficiency.
Patients with 17-hydroxylase deficiency do not manifest symptoms of adrenal insufficiency because of increased production of corticosterone, a glucocorticoid. Because corticosterone is a weaker glucocorticoid than cortisol, very high levels of corticosterone are necessary before feedback inhibition on pituitary ACTH production occurs. As a result, a new steady state is established, with dramatically elevated levels of steroid intermediates, such as progesterone, DOC, and corticosterone.
As the biosynthetic pathway diagram above shows, 17-hydroxylase is not required for aldosterone synthesis. [10] However, the elevated DOC levels from the zona fasciculata result in salt retention, volume expansion, hypertension, hypokalemia, and down-regulation of the renin-angiotensin axis. This secondarily inhibits aldosterone production, which typically is virtually absent in affected patients.
The persistently elevated ACTH levels continue to drive overproduction of the preceding precursors, especially progesterone, DOC, and corticosterone. [11] In these patients, DOC is principally under the control of ACTH rather than angiotensin, and it is predominantly secreted by the zona fasciculata rather than by the glomerulosa. DOC is metabolized in the liver to tetrahydrodeoxycorticosterone, which is then conjugated to glucuronic acid and excreted in the urine. DOC can be further hydroxylated to 19-nordeoxycorticosterone, which also is a potent mineralocorticoid. Thus, 19-Nor-deoxycorticosterone levels also are elevated in patients with this syndrome.
In all variants of 17-hydroxylase deficiency, the production of sex steroids is absent, resulting in a compensatory increase in levels of follicle-stimulating hormone and luteinizing hormone comparable to that in menopause. In humans, the gene product for 17-alpha hydroxylase (P450C17) is expressed in the adrenal cortex, testes, and ovaries but not in the placenta. The adrenals produce glucocorticoids, mineralocorticoids, and C-19 steroids. The gonads, on the other hand, predominantly produce the C-19 steroids and sex hormones. Thus, in patients with 17-hydroxylase deficiency, adrenal and gonadal steroidogenesis are impaired.
P450C17 performs multiple biochemical transformations. It 17-hydroxylates pregnenolone and progesterone and also is responsible for 17,20-lyase activity. Lin and colleagues described the differential regulation of the 2 principal activities of P450C17. [12] Also, Zhang and associates described the developmentally regulated expression of P450C17. [13] They suggested that P450C17 may play an important role in adrenarche, an event in children that is characterized by a dramatic rise in adrenal dehydroepiandrosterone (DHEA) production.
Patients with 17-hydroxylase deficiency typically have impairments of 17-alpha-hydroxylase and 17,20-lyase activity. However, cases of isolated 17,20-lyase deficiency have been described, with CYP17 gene mutations for such cases having been confirmed by molecular genetic studies. [14] Cases of isolated 17-alpha-hydroxylase deficiency have also been described.
Etiology
The human CYP17 gene codes for the 508 amino acid enzyme, which has a molecular weight of approximately 57,000.
-
The enzyme has 17,20-lyase and 17-alpha-hydroxylase activities. [14]
-
The gene consists of 8 exons.
Most of the described clinical cases of 17-hydroxylase deficiency are associated with small base substitutions or insertions, resulting in premature peptide termination, on the gene located in the 10q24-25 band. Single or multiple codon deletions, as well as large deletions, nonsense mutations, and missense mutations, have also been described. [15] Overall, about 40 different mutations of the gene have been described so far. No strong genotype-phenotype correlation exists in 17-hydroxylase deficiency.
As with other variants of congenital adrenal hyperplasia, 17-hydroxylase deficiency is an autosomal recessive disease.
Most mutations are random and spontaneous.
-
Clusters of patients with the same mutation have been identified in Holland, Brazil, and Japan, suggesting a founder effect.
-
Mutations that retain partial enzymatic activity also have been described.
-
A very rare variant featuring combined CYP21A2 and CYP17 deficiency has been described; it appears to result from mutations in the gene for P450 oxidoreductase rather than from mutations in either the CYP17 or CYP21A2 genes.
Epidemiology
United States statistics
The occurrence of 17-hydroxylase deficiency reportedly is very rare. It is responsible for less than 1% of all cases of congenital adrenal hyperplasia. At least 14 cases of isolated 17,20 lyase deficiency have been reported in the presence of normal 17-alpha-hydroxylase activity.
International statistics
A deficiency of 21-hydroxylase is by far the most common variant of CAH (95% of all cases), but the exact prevalence of 17-hydroxylase deficiency is unknown. Most authorities indicate that it is rare and is certainly less common than 11-beta-hydroxylase deficiency. More than 120 cases of C-17 hydroxylase deficiency have been reported in the world medical literature. Prevalence may be more common in Brazil, where there appears to be a founder effect, with more than 80% of the gene mutations identified being due to 2 specific mutations. [16] In Japan, several cases of 17-alpha hydroxylase deficiency associated with elevated aldosterone levels have been reported. The exact pathophysiologic mechanism for this enigmatic situation is unclear.
Race-, sex-, and age-related demographics
Race
Fewer than 150 well-validated cases of 17-hydroxylase deficiency have been documented in the medical literature; therefore, making any definitive statement as to the relative frequency of this condition among the major ethnic groups is difficult.
Cases from all over the world have been described, but a relatively low rate of this syndrome and of congenital adrenal hyperplasia in general appears among Blacks from Africa and from the Black diaspora.
Sex
Because 17-hydroxylase deficiency is an autosomal recessive disease, males and females are affected equally.
It is important to note is that if karyotypes are not checked, the disease will be detected more often in females than in males, because males with classic 17-hydroxylase deficiency are phenotypic females.
Age
The most common stage of life at which 17-hydroxylase deficiency is detected is late adolescence, when the lack of sexual development associated with the syndrome becomes evident. Patients also may present with hypertension and hypokalemia. [17] In contrast to 21-hydroxylase deficiency, 17-hydroxylase deficiency is not identified by newborn screening. [18]
Prognosis
Morbidity/mortality
Mortality and major morbidities associated with 17-hydroxylase deficiency stem mainly from delayed recognition or nonrecognition of hypertension.
The long-term sequelae of myocardial infarction, cerebrovascular accident, renal failure, heart failure, and peripheral vascular disease may occur if blood pressure is not well controlled.
The psychologic impact of the hypogonadism and/or ambiguous genitalia associated with the condition may impact the quality of life and social adjustment of patients with 17-hydroxylase deficiency.
Complications
Complications related to hypergonadotropic hypogonadism are as follows:
-
Delayed epiphysial fusion resulting in eunuchoid habitus and tall adult stature
-
Dyslipidemia
Complications related to chronic hypertension are as follows:
-
Cerebrovascular accidents
-
Acute coronary syndromes
Iatrogenic Cushing syndrome
-
Generic adrenocortical steroidogenesis pathway.
-
Adrenal steroidogenesis pathway (same as synthetic pathway in gonads).






