Practice Essentials
Brugada syndrome is a disorder characterized by sudden death associated with one of several ECG patterns characterized by incomplete right bundle-branch block and ST-segment elevations in the anterior precordial leads. See the image below.
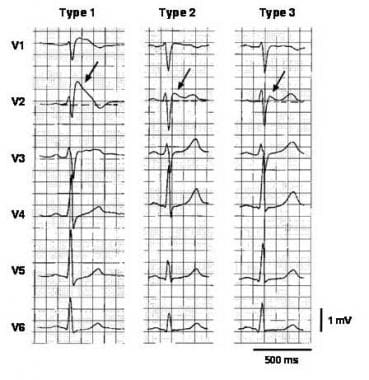 Three types of ST-segment elevation in Brugada syndrome, as shown in the precordial leads on ECG in the same patient at different times. Left panel shows a type 1 ECG pattern with pronounced elevation of the J point (arrow), a coved-type ST segment, and an inverted T wave in V1 and V2. The middle panel illustrates a type 2 pattern with a saddleback ST-segment elevated by >1 mm. The right panel shows a type 3 pattern in which the ST segment is elevated < 1 mm. According to a consensus report (Antzelevitch, 2005), the type 1 ECG pattern is diagnostic of Brugada syndrome. Modified from Wilde, 2002. Image courtesy of Richard Nunez, MD, and EMedHome.com (http://www.emedhome.com/).
Three types of ST-segment elevation in Brugada syndrome, as shown in the precordial leads on ECG in the same patient at different times. Left panel shows a type 1 ECG pattern with pronounced elevation of the J point (arrow), a coved-type ST segment, and an inverted T wave in V1 and V2. The middle panel illustrates a type 2 pattern with a saddleback ST-segment elevated by >1 mm. The right panel shows a type 3 pattern in which the ST segment is elevated < 1 mm. According to a consensus report (Antzelevitch, 2005), the type 1 ECG pattern is diagnostic of Brugada syndrome. Modified from Wilde, 2002. Image courtesy of Richard Nunez, MD, and EMedHome.com (http://www.emedhome.com/).
Signs and symptoms
Signs and symptoms in patients with Brugada syndrome may include the following:
-
Syncope and cardiac arrest: Most common clinical manifestations; in many cases, cardiac arrest occurs during sleep or rest
-
Nightmares or thrashing at night
-
Asymptomatic, but routine ECG shows ST-segment elevation in leads V1-V3
-
Associated atrial fibrillation (20%) [1]
-
Fever: Often reported to trigger or exacerbate clinical manifestations
The lack of a prodrome has been reported to be more common in patients with ventricular fibrillation documented as the cause of syncope in patients with Brugada syndrome. [2]
See Presentation for more detail.
Diagnosis
Most patients with Brugada syndrome have a normal physical examination. However, such an examination is necessary to exclude other potential cardiac causes of syncope or cardiac arrest in an otherwise healthy patient (eg, heart murmurs from hypertrophic cardiomyopathy or from a valvular or septal defect).
Testing
In patients with suspected Brugada syndrome, consider the following studies:
-
12-lead ECG in all patients with syncope
-
Drug challenge with a sodium channel blocker in patients with syncope without an obvious cause
-
Electrophysiologic study to determine the inducibility of arrhythmias for risk stratification
Laboratory tests that may aid in the diagnosis of Brugada syndrome include the following:
-
Serum potassium and calcium levels: In patients presenting with ST-segment elevation in the right precordial leads
-
Potassium and calcium levels: ECG patterns in patients with hypercalcemia and hyperkalemia similar to that of Brugada syndrome
-
CK-MB and troponin levels: In patients with symptoms compatible with an acute coronary syndrome
-
Genetic testing for a mutation in SCN5A
Further testing may be indicated to exclude other diagnostic possibilities.
Imaging studies
Perform echocardiography and/or MRI, primarily to exclude arrhythmogenic right ventricular cardiomyopathy, as well as to assess for other potential causes of arrhythmias.
See Workup for more detail.
Management
To date, the only treatment that has proven effective in treating ventricular tachycardia and fibrillation and preventing sudden death in patients with Brugada syndrome is implantation of an automatic implantable cardiac defibrillator (ICD). Radiofrequency catheter ablation has been recently reported as an effective new treatment. [3, 4, 5, 6, 7]
No pharmacologic therapy has been proven to reduce the occurrence of ventricular arrhythmias or sudden death; however, theoretically, drugs that counteract the ionic current imbalance in Brugada syndrome could be used to treat it. For example, quinidine, which blocks the calcium-independent transient outward potassium current (Ito), has been shown to normalize the ECG pattern in patients with Brugada syndrome. [8] However, quinidine also blocks sodium (Na) currents, which could have contrary effects.
See Treatment and Medication for more detail.
Background
Brugada syndrome is a disorder characterized by sudden death associated with one of several electrocardiographic (ECG) patterns characterized by incomplete right bundle-branch block and ST elevations in the anterior precordial leads. See the image below.
Three types of ST-segment elevation in Brugada syndrome, as shown in the precordial leads on ECG in the same patient at different times. Left panel shows a type 1 ECG pattern with pronounced elevation of the J point (arrow), a coved-type ST segment, and an inverted T wave in V1 and V2. The middle panel illustrates a type 2 pattern with a saddleback ST-segment elevated by >1 mm. The right panel shows a type 3 pattern in which the ST segment is elevated < 1 mm. According to a consensus report (Antzelevitch, 2005), the type 1 ECG pattern is diagnostic of Brugada syndrome. Modified from Wilde, 2002. Image courtesy of Richard Nunez, MD, and EMedHome.com (http://www.emedhome.com/).
In the initial description of Brugada syndrome, the heart was reported to be structurally normal, but this concept has been challenged. [9] Subtle structural abnormalities in the right ventricular outflow tract have been reported.
Brugada syndrome is genetically determined and has an autosomal dominant pattern of transmission in about 50% of familial cases (see Etiology). The typical patient with Brugada syndrome is young, male, and otherwise healthy, with normal general medical and cardiovascular physical examinations.
Patients with Brugada syndrome are prone to develop ventricular tachyarrhythmias that may lead to syncope, cardiac arrest, or sudden cardiac death. [10, 11, 12] Infrahisian conduction delay and atrial fibrillation may also be manifestations of the syndrome. [13, 14]
About 5% of survivors of cardiac arrest have no clinically identified cardiac abnormality. About half of these cases are thought to be due to Brugada syndrome. [15]
At present, implantation of an automatic implantable cardiac defibrillator (ICD) is the only treatment proven effective in treating ventricular tachycardia and fibrillation and preventing sudden death in patients with Brugada syndrome (see Treatment).
Pathophysiology
Brugada syndrome is an example of a channelopathy, a disease caused by an alteration in the transmembrane ion currents that together constitute the cardiac action potential. Specifically, in 10-30% of cases, mutations in the SCN5A gene, which encodes the cardiac voltage-gated sodium channel Nav 1.5, have been found. These loss-of-function mutations reduce the sodium current (INa) available during the phases 0 (upstroke) and 1 (early repolarization) of the cardiac action potential.
This decrease in INa is thought to affect the right ventricular endocardium differently from the epicardium. Thus, it underlies both the Brugada ECG pattern and the clinical manifestations of the Brugada syndrome.
The exact mechanisms underlying the ECG alterations and arrhythmogenesis in Brugada syndrome are disputed. [16] The repolarization-defect theory is based on the fact that right ventricular epicardial cells display a more prominent notch in the action potential than endocardial cells. This is thought to be due to an increased contribution of the transient outward current (Ito) to the action potential waveform in that tissue.
A decrease in INa accentuates this difference, causing a voltage gradient during repolarization and the characteristic ST elevations on ECG. Research has provided human evidence for a repolarization gradient in patients with Brugada syndrome using simultaneous endocardial and epicardial unipolar recordings. [17] See the image below.
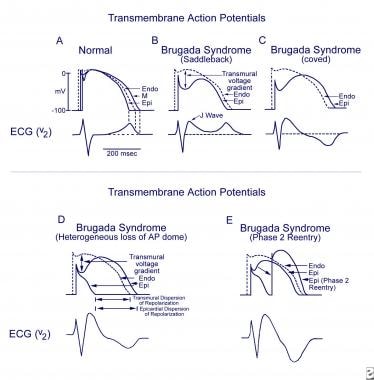 Schematics show the 3 types of action potentials in the right ventricle: endocardial (End), mid myocardial (M), and epicardial (Epi). A, Normal situation on V2 ECG generated by transmural voltage gradients during the depolarization and repolarization phases of the action potential. B-E, Different alterations of the epicardial action potential that produce the ECG changes observed in patients with Brugada syndrome. Adapted from Antzelevitch, 2005.
Schematics show the 3 types of action potentials in the right ventricle: endocardial (End), mid myocardial (M), and epicardial (Epi). A, Normal situation on V2 ECG generated by transmural voltage gradients during the depolarization and repolarization phases of the action potential. B-E, Different alterations of the epicardial action potential that produce the ECG changes observed in patients with Brugada syndrome. Adapted from Antzelevitch, 2005.
When the usual relative durations of repolarization are not altered, the T wave remains upright, causing a saddleback ECG pattern (type 2 or 3). When the alteration in repolarization is sufficient to cause a reversal of the normal gradient of repolarization, the T wave inverts, and the coved (type 1) ECG pattern is seen. In a similar way, a heterogeneous alteration in cardiac repolarization may predispose to the development of reentrant arrhythmias, termed phase 2 reentry, that can clinically cause ventricular tachycardia and ventricular fibrillation. [18]
An alternative hypothesis, the depolarization/conduction disorder model, proposes that the typical Brugada ECG findings can be explained by slow conduction and activation delays in the right ventricle (in particular in the right ventricular outflow tract). [16]
One study used ajmaline provocation to elicit a type 1 Brugada ECG pattern in 91 patients, and found that the repolarization abnormalities were concordant with the depolarization abnormalities and appeared to be secondary to the depolarization changes. [19] Using vectorcardiograms and body surface potential maps, investigators were able to show that depolarization abnormalities and conduction delay mapped to the right ventricle.
Etiology
The prototypical case of Brugada syndrome has been associated with alterations in the SCN5A gene, of which nearly 300 mutations have been described. [20] Mutations in other genes have been proposed to cause a variant of Brugada syndrome, including the genes coding for alpha1- and beta2b-subunits of the L-type calcium channel (CACNA1C and CACNB2), which are thought to cause a syndrome of precordial ST elevation, sudden death, and short QT interval. [21]
Mutations in the genes GPD1-L [22] and SCN1B [23] have been identified in a few familial cases. Cases in which a mutation in the SCN5A gene cannot be demonstrated may be due to mutations of these genes, due to other unidentified genes, or located in regions of the coding sequence or promoter region of SCN5A that are not routinely sequenced in lab tests.
Many clinical situations have been reported to unmask or exacerbate the ECG pattern of Brugada syndrome. Examples are a febrile state, hyperkalemia, hypokalemia, hypercalcemia, alcohol or cocaine intoxication, and the use of certain medications, including sodium channel blockers, vagotonic agents, alpha-adrenergic agonists, beta-adrenergic blockers, heterocyclic antidepressants, and a combination of glucose and insulin. [18]
Epidemiology
United States statistics
Because of its recent identification, the prevalence of Brugada syndrome is not well established. In a large university hospital on the West Coast of the United States, the prevalence of a Brugada ECG pattern among unselected, mainly white and Hispanic adults was 2 of 1348 patients (0.14%); in both cases, the ECG patterns were type 2. [24] The prevalence in Asian and other ethnic populations may be higher.
International statistics
The highest prevalence of Brugada syndrome is in Southeast Asia; the lowest is in North Africa. [25] In parts of Asia (eg, the Philippines, Thailand, Japan), Brugada syndrome seems to be the most common cause of natural death in men younger than 50 years. It is known as Lai Tai (Thailand), Bangungot (Philippines), and Pokkuri (Japan). In Northeast Thailand, the mortality rate from Lai Tai is approximately 30 cases per 100,000 population per year. [26]
Race-, sex-, and age-related demographics
Brugada syndrome is most common in people from Asia. The reason for this observation is not yet fully understood but may be due to an Asian-specific sequence in the promoter region of SCN5A. [27]
Brugada syndrome is 8-10 times more prevalent in men than in women, although the probability of having a mutated gene does not differ by sex. The penetrance of the mutation therefore appears to be much higher in men than in women.
Brugada syndrome most commonly affects otherwise healthy men aged 30-50 years, but affected patients aged 0-84 years have been reported. The mean age of patients who die suddenly is 41 years. [18]
Prognosis
Brugada syndrome is a cause of polymorphic ventricular tachycardia, which may degenerate into ventricular fibrillation and cause cardiac arrest. Prolonged hypoxia during cardiac arrest may leave patients with neurologic sequelae. Implantable cardioverters-defibrillators (ICDs) are often used to treat patients with Brugada syndrome, exposing them to complications related to device implantation and the potential for inappropriate shocks.
During a mean follow-up of 24 months, sudden cardiac death or ventricular fibrillation occurred in 8.2% of patients with Brugada syndrome. A history of syncope, a spontaneously abnormal ECG, and inducibility during programmed electrical stimulation (by one study) significantly increased this risk. [11]
Brugada syndrome may be a significant cause of death, aside from accidents, in men under 40. The true incidence is not known due to reporting biases. Although there is a strong population dependence, an estimated 4% of all sudden deaths and at least 20% of sudden deaths in patients with structurally normal hearts are due to the syndrome. Those with the syndrome have a mean age of sudden death of 41 ±15 years. [28]
Patient Education
Educating the patient and his or her family members and coworkers about basic cardiopulmonary resuscitation (CPR) is important. Genetic counseling is reasonable if desired by the patient and family.
-
Schematics show the 3 types of action potentials in the right ventricle: endocardial (End), mid myocardial (M), and epicardial (Epi). A, Normal situation on V2 ECG generated by transmural voltage gradients during the depolarization and repolarization phases of the action potential. B-E, Different alterations of the epicardial action potential that produce the ECG changes observed in patients with Brugada syndrome. Adapted from Antzelevitch, 2005.
-
Three types of ST-segment elevation in Brugada syndrome, as shown in the precordial leads on ECG in the same patient at different times. Left panel shows a type 1 ECG pattern with pronounced elevation of the J point (arrow), a coved-type ST segment, and an inverted T wave in V1 and V2. The middle panel illustrates a type 2 pattern with a saddleback ST-segment elevated by >1 mm. The right panel shows a type 3 pattern in which the ST segment is elevated < 1 mm. According to a consensus report (Antzelevitch, 2005), the type 1 ECG pattern is diagnostic of Brugada syndrome. Modified from Wilde, 2002. Image courtesy of Richard Nunez, MD, and EMedHome.com (http://www.emedhome.com/).
Tables
Characteristic |
Type 1 |
Type 2 |
Type 3 |
J wave amplitude |
≥2 mm |
≥2 mm |
≥2 mm |
T wave |
Negative |
Positive or biphasic |
Positive |
ST-T configuration |
Cove-type |
Saddleback |
Saddleback |
ST segment, terminal portion |
Gradually descending |
Elevated by ≥1 mm |
Elevated by < 1 mm |





