Practice Essentials
Abnormalities in the coagulation cascade that are independent of platelet mechanisms can affect hemostasis. These abnormalities may be inherited or acquired.
Disorders of nonplatelet hemostasis can be divided into 2 groups based on whether they increase or decrease coagulation. The former may lead to thrombosis, the latter to hemorrhage. Such a division is not absolute, since some disorders may have both hemorrhagic and thrombotic manifestations.
Coagulation-promoting conditions include the following:
-
Procoagulant afibrinogenemia/ dysfibrinogenemia
-
Activated protein C resistance (aPCR)
Coagulation-impeding conditions include the following:
-
Anticoagulant afibrinogenemia/dysfibrinogenemia
-
Factor V deficiency
-
Factor XII deficiency
-
Hypoprothrombinemias
Etiologically, nonplatelet hemostatic disorders can be divided those involving coagulation factors and those involving vascular aspects of hemostasis. See the image below.
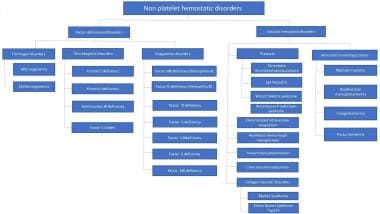 Approach to classification of nonplatelet hemostatic disorders.
Approach to classification of nonplatelet hemostatic disorders.
The Coagulation Cascade
Hemostasis is a dynamic and tightly regulated process, with primary hemostasis involving formation of a platelet plug and secondary hemostasis pertaining to activation of the clotting cascade and ultimately the formation of a clot. [1] Blood coagulation is triggered by the exposure of tissue factor at injury sites and its interaction with activated factor VII, leading to the generation of minute quantities of thrombin. [2]
Thrombin, in turn, activates platelets, as well as factors XI, VIII, and V, and triggers the sequential activation of factors XI, IX, X, and prothrombin on the activated platelet surface, leading to the amplification of thrombin generation to sufficient amounts to convert fibrinogen to fibrin. Endothelial cells have also been demonstrated to have a crucial role as binding sites for various clotting factors at the sites of injury. [3] Platelets localize coagulation to the hemostatic thrombus and protect coagulation enzymes from inhibition by plasma and platelet inhibitors, thus preventing disseminated intravascular coagulation (DIC).
Mechanisms are also in place that limit the hemostatic response to the site of injury and prevent further platelet activation. Those include production of nitric oxide and prostacyclin (PGI2) as well as CD39 from endothelial cells. [3]
Fibrinogen Disorders
Fibrinogen disorders can be quantitative (afibrinogenemia) or qualitative (dysfibrinogenemia), and they may be congenital or acquired. Afibrinogenemia is defined as a deficiency or absence of fibrinogen (coagulation factor I) in the blood. Dysfibrinogenemias involve structural defects in fibrinogen that alter the conversion of fibrinogen to fibrin.
Approximately 300 abnormal fibrinogens have been reported, and about 83 structural defects have been identified. [4] The most common structural defect involves the fibrinopeptides and their cleavage sites; the second most common involves the gamma-chain polymerization region.
Mechanisms of dysfibrinogenemias include the following:
-
Impaired release of fibrinopeptides
-
Defective fibrin polymerization
-
Abnormal cross-linking by activated factor XIIIa (factor XIIIa)
-
Abnormal interactions with platelets
-
Defective fibrinolysis
-
Defective assembly of the fibrinolytic system
-
Abnormal calcium binding
Some patients with dysfibrinogenemia have additional hemostasis defects, including factor V Leiden and deficiencies in antithrombin, protein C, and protein S. [5]
The true prevalence of congenital fibrinogen disorders is unknown. The condition appears to be rare, however, with only 400 families reported as of 2012. [6, 7] Of those patients, 55% were asymptomatic (their cases detected by chance), 25% had bleeding symptoms, and 20% presented with thrombosis. The prevalence of dysfibrinogenemia in patients with a history of venous thrombosis is low (0.8%), as deduced from 9 studies in 7 countries on 2376 subjects. [7] No variation by race, age, or sex is known. Mortality is related to the severity of bleeding and/or to thrombotic complications at presentation.
Acquired fibrinogen deficiency can be associated with several clinical conditions including excessive blood loss in the setting of surgery, trauma or postpartum hemorrhage, hemodilution from from massive transfusion, liver disease, increased consumption in the setting of cancer, DIC or tPA therapy, or assay interference with direct or other thrombin inhibitors. [8]
Presentation
While most patients with dysfibrinogenemia are clinically asymptomatic, some present with a bleeding diathesis, others with thrombophilia, and others with both bleeding and thromboembolism. [5]
Approximately half of the mutations are clinically silent. Hemorrhage and thrombosis occur in almost equal numbers of cases. Severe bleeding is rare and is typically limited to postpartum bleeding, although isolated cases of patients with intra-abdominal hemorrhage presenting with acute abdomen have been reported. [9, 10, 11] Clinical bleeding severity is correlated to the fibrinogen level. [12]
The majority of patients with afibrinogenemia suffer from bleeding events although those are generally less severe than in hemophilia, [13] but thrombotic events may also occur. Dysfibrinogenemias present particular problems for the obstetrician because women affected by these disorders are at increased risk of first-trimester bleeding, spontaneous abortion, and/or postpartum thrombosis. [14] Patients are also high risk for bleeding in surgery.
The diagnosis of afibrinogenemia/dysfibrinogenemia should be considered in a patient who has bleeding or thrombosis unexplained by other common causes. A high level of clinical suspicion should be maintained in patients with other inherited disorders of hemostasis, such as protein C or S deficiency.
Diagnosis
The laboratory diagnosis of dysfibrinogenemias is difficult. Considerations include the following:
-
Fibrinogen antigen level is preserved but activity is markedly decreased. [15]
-
Prothrombin time (PT) appears to be more sensitive than activated partial thromboplastin time (aPTT).
-
Thrombin time (TT) and reptilase time (RT) are typically prolonged. [16]
-
Fibrinogen levels are usually less than 100 mg/dL in the absence of iatrogenic causes (eg, massive blood loss, antifibrinolytics agents).
-
Screening test results (eg, PT, aPTT) may be within reference ranges or only slightly prolonged. [17]
-
Fibrinogen levels are decreased with DIC, primary and secondary fibrinolysis, and liver disease.
Because fibrinogen is an acute-phase protein reactant, increased levels may be observed with inflammation. Pregnancy and oral contraceptive use may also increase plasma fibrinogen levels. Plasma fibrinogen levels vary between the sexes and with weight, glucose levels, triglyceride values, and low levels of high-density lipoprotein cholesterol in healthy adults. [18]
Because of all these variations, many clinicians consider measurement of fibrinogen activity by thromboelastography (TEG) or thromboelastometry to be the most accurate measurement of dysfibrinogenemia or qualitative dysfunctions. [19, 20, 21] These assays evaluate clot strength and firmness, with elastometry showing a better association with the Clauss method of measuring fibrinogen levels. [21] The two assays are highly correlated but not interchangeable. [8]
Treatment
Treatment of afibrinogenemia/dysfibrinogenemia depends on the presenting clinical setting. Plasma fibrinogen is best replaced by cryoprecipitate. Purified, virally inactivated fibrinogen concentrates (eg, RiaSTAP) can be used if available. [22, 23]
Prophylactic administration of blood product or fibrinogen therapy has no role. Recommendations for a desired plasma fibrinogen level for specific conditions are difficult to find because most physicians rely on correction of the clinical hemostatic abnormality as an endpoint. Thromboelastography and thromboelastometry are not recommended to be used in the guidance for transfusion, because although transfusion guided by these tests has been shown to decrease bleeding, it does not decrease morbidity or mortality. [24]
Hunt et al make a practical recommendation of supplementing fibrinogen when levels fall below 1.5 g/L in the setting of massive hemorrhage. [24] Kobayashi et al recommend that the fibrinogen level must be at least 0.60 g/L and, if possible, more than 1 g/L during pregnancy in patients with congenital afibrinogenemia. These authors maintain a plasma level of 150-200 g/dL during labor to prevent placental abruption.
Vascular and Nonvascular Hemostatic Disorders
The following disorders have been classified as nonplatelet vascular and nonvascular hemostatic disorders, although they rarely manifest as significant bleeding or thrombotic problems [25] :
-
Hyperglobulinemic purpura
-
Cavernous hemangioma
-
Hereditary hemorrhagic telangiectasia
-
Pseudoxanthoma elasticum
-
Shwartzman phenomenon
-
Disseminated intravascular coagulation
-
Waterhouse-Friderichsen syndrome
Abnormal circulating protein–related disorders
Abnormal circulating proteins may precipitate in the microvasculature, leading to localized thrombosis. These thromboses may be monoclonal, such as those produced in multiple myeloma and Waldenström macroglobulinemia, or they may be polyclonal, such as those found in the cryoglobulinemias. [26] Abnormal circulating proteins are associated with infectious, autoimmune, and neoplastic disorders. Patients typically present with purpuric skin rash, urticaria, arthralgia, motor-sensory polyneuropathy, and diffuse proliferative glomerulonephritis. Laboratory findings may indicate anemia, presence of rheumatoid factor, and decreased complement levels, as well as abnormal populations of paraproteins and/or immunoglobulins. [27, 28]
Purpuras
Purpuras form another group of vascular hemostatic disorders. Hyperglobulinemic purpura caused by increased gamma-globulin levels has a presentation similar to those of the disorders described above. [29]
Immunoglobulin A vasculitis (IgAV), previously known as Henoch-Schönlein purpura (HSP), is a form of nonthrombocytopenic purpura due to hypersensitivity vasculitis that is primarily observed in children. [30, 31] The condition is usually benign, and it manifests in a variety of clinical symptoms, including urticaria and erythema, arthropathy and arthritis, gastrointestinal problems, and kidney involvement. GI involvement can manifest as acute GI bleeding and can be severe. [32] On rare occasions, patients have life-threatening hemorrhage that requires blood and blood-product support. [33]
Soluble urine transferrin receptor as well as the urinary Fc fragment of IgA receptor (CD89) and transglutaminase-2 (TG-2) have been proposed a potential marker for active IgAV nephritis, previously termed HSP nephritis. [34, 32, 35] Diagnosis of IgAV/HSP is based on the European League Against Rheumatism/Paediatric Rheumatology International Trials Organisation/Paediatric Rheumatology European Society (EULAR/PReS/PRINTO) criteria with the mandatory presence of purpura or petechiae with lower extremity predominance and 1 out of 4 of the following:
- Acute-onset diffuse abdominal colicky pain (may include intussusception and gastrointestinal bleeding)
- Histology showing leukocytoclastic vasculitis or proliferative glomerulonephritis with predominant IgA deposition
- Acute onset arthralgia or arthritis
- Either proteinuria or hematuria
Kidney biopsy is warranted for severe proteinuria, persistent moderate proteinuria or impaired glomerular filtration rate.
Most cases of IgAV improve spontaneously. Treatment is otherwise directed by the specific organ involvement. [32]
Waterhouse-Friderichsen syndrome
Waterhouse-Friderichsen syndrome is characterized by the abrupt onset of fever, petechiae, arthralgia, weakness, and myalgias, followed by acute hemorrhagic necrosis of the adrenal glands and severe cardiovascular dysfunction. The syndrome is most often associated with meningococcal septicemia but may complicate sepsis caused by other organisms, including certain streptococcal species. This disorder may be associated with a history of splenectomy. [36]
Wiskott-Aldrich syndrome
Wiskott-Aldrich syndrome (WAS) is a rare X-linked immunodeficiency syndrome characterized by eczema, thrombocytopenic purpura, and recurrent pyogenic infection. The syndrome is observed exclusively in young boys. Typically, immunoglobulin M levels are low and immunoglobulin A and E levels are elevated. Lymphoreticular malignancies are common. [37]
WAS can present with a wide clinical spectrum. When the diagnosis is suspected, screening with flow cytometry analysis of WAS protein (WASp) expression is warranted and further testing is indicated if screening is positive. Once the diagnosis is made, a clinical score can be assigned based on clinical features, with a score of 3 or higher corresponding to "classic" WAS. Management of these patients involves a multidisciplinary approach, with hematopoietic stem cell transplantation being the only curative approach. [38]
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura (TTP) is a clinical syndrome classically characterized by neurologic symptoms, fever, renal impairment, thrombocytopenia, hemolytic anemia, and microvascular thrombosis that result in variable degrees of tissue ischemia and infarction. Large-vessel thrombosis is uncommon.
The pathogenesis of TTP involves a relationship between the actions of ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif, 13) and platelet adherence to the extracellular vascular matrix. ADAMTS13 is a plasma zinc metalloprotease that cleaves von Willebrand factor (vWF) in the process of coagulation. A deficiency of ADAMTS13 creates a propensity for increased vWF-platelet aggregation that results in the intravascular thrombosis seen in TTP.
While levels of ADAMTS13 are very helpful in a definitive diagnosis, assay results take days in most centers and should not be waited upon to start plasmapheresis. In a clinically suggestive picture, elevated lactate dehydrogenase (LDH) and the presence of schistocytes on peripheral smear are sufficient starting points for emergent treatment. Diagnostic criteria include presence of a microangiopathic hemolytic anemia plus thrombocytopenia, which does not exclude other diagnoses. [39]
ADAMTS13 deficiency can be caused by a genetic mutation or acquired autoimmune inhibitors. Hereditary TTP is a rare autosomal recessive disorder caused by ADAMTS13 mutations that result in the absence or severe deficiency of the plasma metalloprotease ADAMTS13. Patients with hereditary TTP may appear to be healthy, but their increased risk of critical thrombosis is always present. [40] More than 200 ADAMTS13 mutations, spread over all ADAMTS13 protein domains, have been identified in patients with hereditary TTP. Diagnosis of hereditary TTP is confirmed by severely deficient ADAMTS13 activity (≤10% of normal) in the absence of a functional inhibitor and the presence of ADAMTS13 mutations on both alleles. [41]
In patients with thrombocytopenia and schistocytes, the Plasmic score can be used to predict ADAMTS13 level less than 10%. [42]
Clinical features of the disease vary, with some patients having minimal findings and others being critically ill. Most patients have normal or only transiently increased serum creatinine. [39]
Several drugs have been implicated in the development of inhibitors and clinical TTP, including cyclosporine, mitomycin-C, ticlopidine, simvastatin, atorvastatin (Lipitor), and clopidogrel (Plavix). Human immunodeficiency virus (HIV) infection has also been associated with TTP.
Therapeutic daily plasma exchange with 40 mL fresh frozen plasma (FFP)/kg of body weight is the treatment of first choice in acute TTP and thrombotic thrombocytopenic purpura–adult hemolytic uremic syndrome (TTP-HUS) until complete remission. FFP replenishes the deficient ADAMTS13, while plasma exchange removes some of the pathogenic autoantibodies and endothelial-stimulating cytokines. Octaplas—a pooled plasma (human) blood product that has been treated with a solvent detergent process—provides a viable alternative to single-donor FFP and provides a reduced risk of certain viral transmissions. Twice-daily plasma exchange has been used as salvage therapy in severe cases. [43]
McCarthy et al have treated more than 160 patients using FFP, solvent detergent (SD), and cryosupernatant as the exchange media. [44] They showed that SD plasma has value in virtually eliminating all allergic reactions during treatment. Approximately 80% of patients respond to plasma exchange therapy.
For patients with severe plasma allergic reactions, treatment with plasma-derived factor VIII concentrate that contains ADAMTS13 is an option. [39] Corticosteroids are routinely added for the management of an acute episode, although high-quality evidence-based studies for the use of steroids are lacking. [45]
Rituximab, a monoclonal antibody against CD20 present in B-lymphoid cells, has been successfully used in treating patients with acquired TTP who had failed to respond to plasma exchange. It is also used preemptively in patients with a persistent or recurrent ADAMTS13 deficiency. [45]
Caplacizumab, an anti-vWF humanized, bivalent variable-domain-only immunoglobulin fragment, inhibits interaction between vWF multimers and platelets and has shown efficacy in the treatment of acquired TTP. However, a subgroup of patients who had persistent ADAMTS13 deficiency had a relapse soon after treatment with caplacizumab was stopped, which suggests that monitoring of ADAMTS13 could be useful to guide the continuation of therapy. [46]
In the phase III HERCULES trial to study the efficacy and safety of caplacizumab in patients with acquired TTP, caplacizumab was associated with faster normalization of the platelet count; a lower incidence of a composite of TTP-related death, recurrence of TTP, or a thromboembolic event during the treatment period; and a lower rate of recurrence of TTP during the trial than placebo. [47]
Other immunomodulatory drugs that are used as third-line options in refractory TTP include vincristine, cyclosporine, cyclophosphamide, and mycophenolate mofetil. [45]
Severe ADAMTS13 deficiency is suggestive of hereditary TTP and identifies a subgroup of patients who respond well to plasma exchange. High-titer ADAMTS13 inhibitors correlate strongly with a high risk of relapsing disease. Knovich et al have developed an ADAMTS13 assay suitable for guiding the treatment of patients with suspected TTP. [48] Recombinant ADAMTS13 may provide specific and more efficacious treatment of patients with TTP. Antiplatelet agents to inhibit the interaction between vWF and platelets are also being studied. [49]
In a review of membrane plasma exchange for the treatment of idiopathic TTP, Marn Pernat et al reported that 52 of 56 patients (93%) had an excellent response to treatment, with 48 (86%) experiencing complete remission (platelet count > 100 x 109/L). The patients (36% of whom were found to have renal impairment) underwent membrane plasma exchange 1-2 times daily until their platelet count normalized, with 1-1.5 plasma volumes (3606 +/- 991 mL) being replaced with FFP during each procedure. Overall, 1066 plasma exchange procedures were performed in this group of patients, with each patient undergoing an average of 19 +/- 17 procedures. [50]
Four patients died after having received only 1-3 procedures; 6 patients who had had a complete remission suffered 1-5 relapses annually during the follow-up period, 1 of whom died of acute hemolytic reaction while undergoing tapering of plasma exchange procedures. In addition, 3 patients required additional splenectomy. The authors concluded that the use of plasma exchange with FFP "as a mandatory, up-to-date therapy" is supported by their data. [50]
Recombinant ADAMTS13 is being investigated as a potential treatment option in patients with hereditary TTP. [43]
Heritable connective-tissue abnormalities and/or vascular malformations
A third group of vascular hemostatic disorders includes those associated with heritable connective-tissue abnormalities and/or vascular malformations. [51] These disorders are considered hemostatic, in part because of their predilection for bleeding or thrombosis and for the development of consumptive coagulopathies after either hemorrhage or excision. [52]
Two inherited connective-tissue disorders have major cardiovascular complications: Marfan syndrome and Ehlers-Danlos syndrome type IV. [53]
Marfan syndrome results from mutations in the FBN1 gene, which encodes fibrillin-1, an extracellular matrix component found in structures called microfibrils. Ehlers-Danlos syndrome type IV results from mutations in the COL3A1 gene, which encodes the polypeptides in type III collagen.
Marfan syndrome remains primarily a clinical diagnosis. Biochemical analysis of the amount of type III collagen produced by dermal fibroblasts has proven to be a powerful diagnostic test for Ehlers-Danlos syndrome type IV. The most common manifestations of Ehlers-Danlos syndrome are hyperextensible skin and joints, skin fragility, and reduced wound-healing capability. Collagen disorders are associated with congenital intracranial aneurysms, accounting for approximately 5% of these cases. [54] Patients with Ehlers-Danlos syndrome may present with sudden, massive gastrointestinal hemorrhage. [55]
Freeman and colleagues reported 95 complications from Ehlers-Danlos type IV syndrome.Their series included 45 subjects with vascular problems, including 22 with spontaneous intra-abdominal hemorrhage. [56] They recommend treatment with nonoperative (ie, angiographic) interventions as a first step, followed by simple vessel ligation.
Cavernous hemangiomata and hereditary hemorrhagic telangiectasias can be loosely added to this group. Cavernous hemangiomata are vascular tumors composed of large dilated blood vessels, often containing large amounts of blood. They can be found in the brain, skin, subcutaneous tissue, and many abdominal viscera, particularly the liver, spleen, and pancreas. [57]
Hereditary hemorrhagic telangiectasia is an autosomal dominant inherited disease associated with various vascular malformations. [58] The disease is caused by defects of transmembrane protein components of the receptor complex for transforming growth factor-beta (TGF-beta). Vascular malformations can be found in the pulmonary, spinal, intracerebral, and hepatic circulation. They vary in size and may cause no symptoms, or they may be responsible for hemorrhage, thrombosis, cardiac insufficiency, portal hypertension, and hepatic encephalopathy secondary to shunting. Hepatic involvement can usually be confirmed with color duplex ultrasonography. Embolization or ligation of the malformations is the main therapeutic strategy.
Shwartzman phenomenon
The Shwartzman phenomenon is defined as local or systemic vasculitis caused by a 2-stage reaction. An initial exposure to endotoxin produces intravascular fibrin thrombi whose clearance results in reticuloendothelial blockade. This prevents the clearance of thrombi generated by a second exposure to endotoxin, polyanions, glycogen, or antigen/antibody complexes, which leads to tissue necrosis and/or hemorrhage.
In pregnancy, gram-negative septicemia during delivery or abortion may serve as the first or provocative encounter. [59] The Shwartzman phenomenon is often associated with sepsis, the systemic inflammatory response syndrome (SIRS), and DIC.
Protein C, Protein S, Antithrombin III, and Factor V Leiden Deficiencies
Deficiencies of antithrombin III, protein C, and protein S are defined as an absence or a reduced level of the protein, leading to an increased risk for thrombosis. These deficiencies can be congenital or acquired. [60] The former are caused by partial or complete gene deletions, replacements, and rearrangements. [61]
Antithrombin III, protein C, and protein S are all essential components of the coagulation process. All are synthesized by the liver and have a half-life in the range of 4-6 hours. Activated antithrombin III is a major inhibitor of thrombin and factor Xa, with smaller effects on factors IX, XI, and XII. Antithrombin III binds to the endothelial cell surface (proteoglycan heparan sulfate) in the presence of injury. It forms a subendothelial cell matrix that neutralizes thrombin by complexing with it. Antithrombin III serves as a cofactor for exogenous heparin, increasing its activity 1000- to 10,000-fold. [62]
Protein C and S are vitamin K–dependent factors that participate in the thrombomodulin–protein C system. Thrombomodulin and thrombin form a complex on the endothelial cell plasma membrane in response to injury, with activated protein S serving as a cofactor. This complex attracts and binds protein C in the presence of calcium ion to produce activated protein C (aPC). aPC inactivates factors Va and VIIIa, thus halting the coagulation cascade. It also neutralizes plasminogen-activator inhibitor I, thereby facilitating fibrinolysis. Deficiency of the naturally occurring anticoagulant proteins antithrombin III, protein C, and protein S, in addition to aPC resistance due to the factor V Leiden gene mutation, are associated with inherited thrombophilia.
Clinical thrombophilia is defined by one or more of the following [63] :
-
An early thromboembolic episode (occurring before age 50 y)
-
Spontaneous thrombosis
-
Recurrent thrombosis
-
Unusual site of thrombosis
-
Family history of thrombotic episodes
-
Warfarin-induced skin necrosis
Such patients may have an isolated or combined inherited deficiency in the proteins involved in coagulation. The diagnosis is confirmed by identification of an isolated or combined inherited coagulant deficiency. All affected patients with inherited thrombophilia are at risk of developing thromboembolic disease ranging from mild, superficial venous thrombosis to lethal pulmonary embolism.
Martinelli et al compared the lifetime probability of developing thrombosis, the type of thrombotic symptoms, and the role of circumstantial triggering factors in 723 first- and second-degree relatives of 150 index subjects with different thrombophilic defects. They found higher risks for thrombosis for subjects with antithrombin (risk ratio [RR], 8.1; 95% confidence interval [CI], 3.4-19.6), protein C deficiency (RR, 7.3; 95% CI, 2.9-18.4), or protein S deficiency (RR, 8.5; 95% CI, 3.5-20.8) compared with those with factor V Leiden (RR, 2.2; 95% CI, 1.1-4.7) or with individuals with normal coagulation. [64]
The risk of thrombosis for subjects with factor V Leiden was lower than that for subjects with any of the 3 other coagulation defects (RR, 0.3; 95% CI, 0.1-1.6), even when arterial and superficial vein thromboses (SVTs) were excluded and the analysis was restricted to deep vein thrombosis (DVT) (RR, 0.3; 95% CI, 0.2-0.5). No association was found between coagulation defects and arterial thrombosis. [64]
The most frequent venous problem was DVT with or without pulmonary embolism—90% in antithrombin III deficiency, 88% in protein C deficiency, 100% in protein S deficiency, and 57% in factor V Leiden—but SVT was also common with factor V Leiden (43%). Approximately 50% of subjects had a predisposing factor for thromboembolism, regardless of which underlying disorder was present. [64]
DeStefano et al looked at the risk of recurrence of thromboembolism in patient with inherited deficiencies in antithrombin, protein C and protein S in the absence of anticoagulation and found that antithrombin deficiency is an independent risk factor for recurrence (hazard ratio [HR] 1.9, 95% CI 1.0-3.9) and that carriers of protein C or S deficiency had a marginal increased risk (HR 1.4, 95% CI 0.9-2.2). [65]
Estimates of the frequency of these defects in a population with venous thrombosis place antithrombin III deficiency at 0.5-4.9%, protein C deficiency at 1.4-8.6%, and protein S deficiency at 1.4-7.5%. [66] Factor V Leiden is the most common disorder and is found in 12-40% of white populations. Protein C and S deficiencies may be more prevalent in Asian populations than in white ones.
Miyata et al identified 54 people with protein C deficiency by screening approximately 26,800 Japanese patients. This represents an observed prevalence of 1 case per 500 patients. [67] These researchers also found that 34 patients with protein C deficiency had earlier onset of acute myocardial infarction and atherothrombotic cerebral infarction compared with healthy patients. Their study suggests that congenital protein C deficiency contributes to an earlier onset of arterial occlusive diseases in Japanese subjects.
A study by Suehisa and colleagues of 113 consecutive Japanese patients with DVT found that 32 (28.3%) were deficient in antithrombin III (1.77%), protein C (7.96%), and protein S (17.7%). In comparison, 10 of the 392 healthy Japanese subjects had protein S deficiency (n = 8, 2.02%) or protein C deficiency (n = 2, 0.5%). The frequency of protein C and S deficiencies in patients with DVT was 15.6 and 7.38 times the control population frequency, respectively, and this difference was statistically significant (P< 0.05). These data suggest that the Japanese population has a high frequency of protein C and S deficiencies. [68]
In Taiwan, Shen and colleagues noted that prothrombin G20210A and factor V Leiden mutations were not found in 113 thrombophilic Chinese patients. [69] Only protein C and S deficiencies were significantly associated with increased risk for the development of thrombosis, with an odds ratio (OR) of 10.6 and 6.7, respectively. These findings suggest that protein C and protein S deficiencies are the most important risk factors for thrombosis in venous thrombophilic patients of Chinese extraction. The true prevalence of these hereditary disorders is unknown because of the high variability of clinical presentation. [70]
Antithrombin III deficiencies take several forms and may be quantitative or qualitative. The following have been described [71] :
-
Loss of the entire molecule
-
Diminution of activity only, with normal concentration
-
Normal activity and concentration but with a decreased sensitivity to heparin
-
Diminished production of antithrombin III
-
Increased loss of antithrombin III
-
Increased consumption of the inhibitor
Antithrombin III deficiency should be considered in any patient who cannot be adequately anticoagulated on heparin or who develops thrombosis while on heparin in the absence of heparin-induced thrombocytopenia. FFP or highly purified concentrates should be administered before starting heparin for patients needing anticoagulation. Adequate antithrombin substitution is lifesaving in patients whose cases of DIC are caused by septic or traumatic shock. Protein C and S deficiencies follow a similar pattern and have similar clinical manifestations.
Factor V has both procoagulant and anticoagulant properties. Activated factor V stimulates the formation of thrombin, whereas anticoagulant factor V acts as a cofactor for aPC in the degradation of factor VIII and factor VIIIa, thereby reducing thrombin formation. High procoagulant factor V levels may enhance prothrombinase activity and increase the risk of thrombosis. Factor V with the Leiden mutation (Arg506Gln) is resistant to aPC lysis compared with the wild type. Resistance to aPC is the most common inherited hypercoagulable state associated with venous thrombosis.
Low anticoagulant factor V levels can reduce aPC cofactor activity in the inactivation of factor VIII (aPCR phenotype), which might also promote thrombosis. Low factor V levels in combination with factor V Leiden could lead to a more severe aPCR phenotype (pseudohomozygous aPCR). [72]
The factor V Q506 mutation is found in 20-60% of white patients with thrombosis. Its prevalence varies between countries; it is highly prevalent (up to 15%) in Scandinavian populations. [73]
In a study of 51 families with factor V Leiden, Samama et al reported that the factor V mutation was present in 84 of 125 family members (81 heterozygous, 3 homozygous). Venous thrombosis was observed in 17 of the 84 family members with the mutation, and in 6 of the 41 with a normal aPCR test finding and no mutation. An associated protein C or protein S deficiency was present in 5 families (10%). [74]
The most frequent clinical manifestations of aPCR or factor V Leiden deficiency are superficial or deep venous thromboembolism (VTE) and/or pulmonary embolism and thrombosis at an unusual site (eg, cerebral, mesenteric, or central retinal vein). A causal relationship is frequently difficult to demonstrate. A precipitating factor was observed in 84% of cases, and a recurrent thrombotic episode occurred in 50% of those affected in Samama et al's study. [74] The risk of thrombosis associated with pregnancy was high in the postpartum period, especially in homozygous women. In homozygous patients, markers of coagulation activation are frequently elevated in those who are untreated. In heterozygous true factor V deficiency, both activity and antigen are about 50% of normal. [75]
Laboratory screening for aPCR is performed by functional tests measuring the effect of aPC on aPTT in plasma containing a heparin neutralizer. Second-generation tests that reduce screening errors are now available. [76] Genetic testing by means of polymerase chain reaction (PCR) for identification of the mutated gene can also be used for the diagnosis of factor V Leiden.
The question of who should be screened remains unsettled. Marz et al have taken an inclusive approach to causes of VTE in describing the interactions between genetic and environmental causes of this disease state. [77] They propose that inborn factors that cause a predisposition to thrombosis are present in most patients who develop VTE.
The relatively rare defects of antithrombin III, protein C, and protein S deficiencies are found in 15-20% of thrombophilic families, in contrast to the common genetic polymorphisms of procoagulant molecules, factor V Leiden, and the prothrombin 20210 A allele. The results of Marz and colleagues' studies of factor V Leiden and prothrombin 20210 A indicate that many symptomatic individuals have more than one (genetic and/or environmental) risk factor. Important nongenetic risk factors include age, tissue damage, oral contraception, pregnancy, obesity, and lack of physical activity.
A thrombophilia workup including the above-discussed laboratory tests is warranted only in young patients with unusual sites of first thrombosis, to decide duration of anticoagulation, use of oral contraceptives, and family counseling. Indiscriminate testing is not warranted.
Disseminated Intravascular Coagulation
DIC is a syndrome characterized by an alteration in the elements involved in blood coagulation due to their use/destruction in widespread blood clotting within the vessels. It may be caused by a wide variety of disorders, including hemorrhage, trauma, sepsis, toxic shock syndrome, endotoxin release, abruptio placentae, and amniotic fluid embolism. [78] Sepsis is the most common cause of DIC.
Etiology
The etiology and progression of DIC are multifactorial and are characterized by defects in the protein C system and in the antithrombin and tissue-factor inhibitor pathways. Tissue factor–dependent activation of coagulation, defective physiological anticoagulant pathways, and impaired fibrinolysis caused by elevated levels of plasminogen activator inhibitor type 1 (PAI-1) can all lead to DIC. Release of tissue factor from endothelial cells or other circulating cells is the most common initiating event. Bacterial factors also release tissue factor as well as proinflammatory and anti-inflammatory cytokines.
Tumor necrosis factor (TNF) and interleukin-8 (IL-8) increase the inflammatory response, while IL-10 inhibits it. IL-1 beta, IL-12, IL-2, granulocyte colony-stimulating factor (G-CSF), and interferon gamma have all been reported to induce coagulation. IL-4, IL-13, and transforming growth factor– beta (TGF-beta) have anticoagulant activity.
These imbalances all promote the development of DIC. Persistence of the triggering agent (eg, a septic locus) leads to a consumption coagulopathy with loss of fibrinogen and platelets and the potential for diffuse bleeding. Failure of the fibrinolytic system elicits deposition of microvascular fibrin and multisystem organ failure (MSOF). [79]
Vervloet and colleagues are proponents of the theory that DIC is an imbalance between coagulation and fibrinolysis mediated by various cytokines and caused by increased levels of PAI-1. [80] Increased levels of PAI-1 produce a procoagulant state characterized by thrombin generation in excess of plasmin and impaired fibrin degradation, leading to widespread fibrin deposition. Thrombin generation proceeds via the extrinsic tissue factor/factor VIIa route simultaneous with consumption of the natural coagulation inhibitors antithrombin III, protein C, and protein S increases. Although levels of plasminogen activator antigen are increased, its activity is almost completely inhibited by PAI-1. High plasma levels of thrombin-antithrombin (TAT) complex can be found.
Growing evidence has shown that damage-associated molecular patterns (DAMPs) are involved in the pathogenesis of DIC. DAMPs including cell- free DNA (cfDNA) and DNA binding proteins are released from dying cells or parenchymal and hematopoietic cells, with cfDNA having been shown to be highly procoagulant, inducing platelet aggregation. [81]
Vervloet and colleagues found that increased PAI-1 levels are associated with poorer outcome and increased severity of multisystem organ failure in patients with DIC from sepsis as well as other causes. Hardaway and Vasquez believe that DIC may be initiated by release of a thrombogenic aminophospholipid from dying tissue or bacterial cells. [82] Coagulation abnormalities secondary to DIC are coupled to the inflammatory response, which aggravates vascular permeability, inflammation, and cell damage in tissues. This combination of events leads to multisystem organ failure and death. DIC may produce acute respiratory distress syndrome through the mechanism of intravascular fibrin formation, vessel occlusion, and localized hypoxia. [83]
In Japan, Watanabe and colleagues measured plasma levels of thrombin-activatable fibrinolysis inhibitor (TAFI) activity and antigen in patients with DIC in a study designed to examine the role of hypofibrinolysis in this disorder. [84] Both TAFI activity and antigen levels were significantly below reference ranges in patients with DIC. Decreases in TAFI activity were inversely correlated with increases in plasma TAT III complex and D-dimer, suggesting that TAFI activity is reduced by thrombin generation and consumption of coagulation factors. TAFI activity levels were not correlated with fibrinogen, plasma alpha2-plasmin inhibitor complex, and tissue plasminogen activator (TPA)/PAI-1 complex levels, thus supporting a role for TAFI as a secondary modulator of fibrinolysis.
DIC is seen in COVID-19 and is strongly associated with fatal outcomes. [85] Elevated levels of pro-inflammatory cytokines have been proposed as one possible mechanism underlying coagulation dysfunction in severe COVID-19. [86]
Epidemiology
Okajima et al examined the incidence, clinical presentation, and underlying disorders associated with DIC. In their series of 1882 subjects suspected of having DIC, 204 were diagnosed with DIC, for an overall incidence of 10.8%. [87] Malignancies led the list of underlying disorders, with 33.8% of subjects having solid tumors and 12.7% having hematologic malignancies. Subjects with aortic aneurysm (10.8%), infections (6.4%), unspecified postoperative complications (4.4%), liver disease (2.9%), obstetric disorders (2.5%), and miscellaneous diseases (26.5%) completed the diverse list.
Clinical manifestations of subjects with DIC varied, depending on underlying disease. The large majority of those with aortic aneurysm (95.5%) or postoperative complications (88.9%) had no clinical signs of DIC. Bleeding was observed in all obstetrical patients and in 32-50% of those with liver disease, hematological malignancies, and solid tumors. Organ failure was observed in up to 33.3% of subjects who had DIC with liver disease, hematological malignancies, and solid tumors. Although all of the subjects with obstetric disorders had bleeding, only 20.0% had organ failure. In contrast, although only 15.4% of subjects with infections had bleeding, 76.9% of these had organ failure. [87]
In a study from Thailand, Chuansumrit et al found a similar broad spectrum of underlying diseases in 100 pediatric patients with DIC. [88] Forty-five subjects were neonates with a mean age of 12.6 days, and 55 were infants, children, and adolescents with a mean age of 6 years and 3 months. Most subjects (91.5%) had complicated underlying conditions, which included congenital anomalies, prematurity, malignancies, hematological disorders, and various diseases. The most commonly found initiator of DIC was gram-negative septicemia. Bleeding and thromboembolic events were found in 59.4% and 19.8% of participants, respectively.
Asakura et al examined the relationship between fibrinolytic enhancement and development of multisystem organ failure in 69 subjects with DIC. [89] Those with both DIC and multisystem organ failure had higher levels of TPA antigen and PAI antigen and more depressed levels of plasma alpha2-plasmin inhibitor complex (PIC) and fibrin/fibrinogen degradation products than those without multisystem organ failure.
A retrospective study from Wuhan, China of 183 consecutive patients with COVID-19 found that 71.4% of patients who did not survive met the criteria for DIC during their hospital stay, compared with just one of the patients (0.6%) who survived. [85]
Diagnosis
The diagnosis of DIC is based on both clinical suspicion and a combination of laboratory test findings. Patients with the following known underlying causes should be carefully observed for indications of the development of DIC (eg, microthrombi, bleeding):
-
Malignancy
-
Trauma
-
Aortic aneurysm
-
Cerebral injury
-
Hepatic surgery
-
Burn injury
-
Hypothermia
-
Massive transfusion
-
Prolonged surgery
The Japanese Society on Thrombosis and Hemostasis released DIC diagnostic criteria in 2017 that include coagulation markers such as soluble fibrin and the thrombin-antithrombin complex. [90]
Evidence of ongoing consumption of coagulation proteins from laboratory testing includes decreasing fibrinogen levels and platelet counts. PT and aPTT may both be prolonged. Peripheral smear may show schistocytes. Increasing plasma levels of D-dimer, fibrinogen split products (FSP), and soluble fibrin monomer (FM), are found as DIC progresses. Elevated D-dimer levels reflect both thrombin and plasmin production. [79] These studies must be repeated to confirm the diagnosis of DIC and to monitor therapeutic progress. [91]
Circulating factors can be used as markers of prognosis in DIC. In 1999, Kotajima et al showed that levels of plasma thrombomodulin, a high-affinity thrombin receptor on vascular endothelial cells, were significantly higher in nonsurvivors of DIC than in survivors (thrombomodulin 3.1+/-1.52 FU/mL vs 8.1+/-3.89 FU/mL). [92] The measurement of antithrombin III has been shown to be a sensitive marker for unfavorable prognosis. [93]
Treatment
The treatment of DIC can be divided into the following components:
- Treatment of the underlying disorder
- Supportive management of bleeding complications
- Treatment aimed at the coagulation process
The triggering underlying disease must be treated aggressively. This may require interventions such as surgical drainage of an abscess or necrotic tissue, antibiotic therapy, control of fever, and volume replacement. Early recognition and treatment of DIC is the key to success, so a high index of clinical suspicion must be maintained.
Continued DIC is characterized by a consumption coagulopathy of platelets. Ongoing bleeding or rapid hemorrhage may lead to anemia. These deficiencies can be corrected by platelet transfusions and administration of cryoprecipitate (to replete fibrinogen) and FFP. A fibrinogen level target of > 100 mg/dL is generally used.
Heparin or low molecular weight heparin may be cautiously used for cases of DIC in which thrombosis predominates, and for prophylaxis in patients with DIC who are critically ill and not bleeding. [78, 79, 94, 95]
The utility of antithrombin concentrates for treatment for DIC remains uncertain, with some studies showing possible benefit in patients with severe infection and DIC. [94] Currently, however, these agents (eg, antithrombin recombinant, antithrombin III) are approved only for use in patients with hereditary antithrombin deficiency.
Recombinant activated human factor VII (rFVIIa) initially showed promise in patients with DIC, postpartum bleeding, sepsis, and postoperative hemorrhage. However, the increased thromboembolic risk has discouraged the use of rFVIIa in this setting. [96, 97]
Recombinant human thrombomodulin alpha (rTM), which acts as an inhibitor of thrombin, was previously shown to have some benfit in thrombosis-predominant patients with DIC, but the SCARLET trial showed no reduction in mortality from the use of thrombomodulin in sepsis-associated coagulopathy. [98]
Coagulation-impairing Deficiencies
Factor V deficiency
Factor V has both procoagulant and anticoagulant properties. Activated factor V stimulates the formation of thrombin, whereas anticoagulant factor V acts as a cofactor for aPC in the degradation of factor VIII/VIIIa, thereby reducing thrombin formation.
An inherited autosomal recessive deficiency of factor V, also known as proaccelerin (or accelerator globulin or labile factor) leads to a rare hemorrhagic tendency known as Owren disease or parahemophilia. The severity of the condition varies from bruising to lethal hemorrhage [99, 100, 101] Both PT and aPTT may be prolonged.
Lak et al identified epistaxis and excessive bleeding after surgery as the most common symptoms in 35 Iranian patients with an inherited factor V deficiency, with plasma levels of 1-10%. [102] More severe symptoms, such as gastrointestinal and central nervous system bleeding, were rare. The severity of bleeding symptoms was only partially related to the degree of factor V deficiency in plasma. Acquired inhibitors of factor V and other defects affecting its storage and processing are rare causes of clinical bleeding, with severity ranging from mild to life threatening.
No factor V concentrate is available yet and FFP remains the mainstay of treatment. For acute bleeding and prophylaxis in planning of an invasive procedure, the goal is to maintain FV levels above 20%. [103] However, a novel plasma-derived factor V concentrate has been developed, and has been shown to correct factor V deficiency in vitro based on normalization of global clotting times and thrombin generation parameters. [104]
Fu et al were successful using a combination of factor replacement, chemotherapy, and plasmapheresis in a patient with spontaneous, life-threatening intracranial bleeding caused by a factor V inhibitor. The patient deteriorated after initial treatment with FFP and platelet transfusions. He was subsequently treated with a combination of plasma exchange and chemotherapy, which led to complete recovery. [105]
Combined deficiency of coagulation factor V and factor VIII is an autosomal recessive disorder observed in a number of populations around the world. However, this disease appears to be most common in the Mediterranean basin, particularly in Jews of Sephardic and Middle Eastern origin living in Israel. [106]
Factor VII deficiency
Factor VII is a vitamin K–dependent glycoprotein essential to the extrinsic pathway of coagulation. Deficiencies may be inherited as an autosomal recessive characteristic or acquired in association with vitamin K deficiency, sepsis, autoantibodies, and inhibitors. [107, 108] The prevalence of congenital deficiency is low, with only 729 individual cases.(involving 221 unique variants) in the factor VII gene described in the Factor VII Gene (F7) Variant Database as of 2019. [109]
The predisposition to bleeding is variable and to some extent depends on the amount of plasma factor VII activity, although this correlation is poor. [110] Menorrhagia and metrorrhagia in females and mucosal bleeding and hemarthrosis in both sexes are the most frequent manifestations. Individuals homozygous for the mutation who have complete absence of factor VII activity in plasma usually die shortly after birth because of severe hemorrhage.
This defect produces prolonged PT, reduced factor VII activity, and normal aPTT. [111] True deficiencies are characterized by very low factor VII activity and low factor VII antigen. Other patients may have normal antigen levels but low activity. [112]
Clinical symptoms and factor VII activity levels in plasma correlate rather poorly. Patients may have prolonged PTs and a mixing study is helpful as rapid, sustained correction suggests a factor VII deficiency, but the final diagnosis is established by quantitative factor VII assays. Treatment consists of rFVIIa, factor replacement with FFP, prothrombin complex concentrates, or plasma-derived factor VII concentrates. RFVIIa is a very useful alternative. Hunault and Bauer have reported several successfully treated patients. [110] Because of the short half-life of factor VIIa, repeated doses must be administered.
Factor X deficiency
Factor X deficiency is a coagulation disorder usually inherited as an autosomal recessive trait, though it can be acquired. This deficiency is characterized by defective activity in both the intrinsic and extrinsic pathways, impaired thromboplastin time, and impaired prothrombin consumption. Factor X circulates as a serine protease that is activated at the point of convergence of the intrinsic and extrinsic coagulation pathways. Activated factor Xa is involved in macromolecular complex formation with its cofactor factor Va, a phospholipid surface, and calcium to convert prothrombin into thrombin. [113] Both PT and aPTT are prolonged.
Factor X deficiency may be acquired in patients with light chain–related amyloidosis. This acquired disorder appears to be secondary to adsorption of factor X to the amyloid fibrils. [114, 115] In 1981, Greipp et al reviewed 30 cases of patients who had amyloidosis with factor X deficiency. [116] Modest deficiency of factor X was often associated with severe bleeding. In many cases, clinical bleeding could not be accounted for by deficiency of factor X alone, leading the authors to believe that coexistent hemostatic defects probably contributed to the bleeding. Testing with Russell viper venom may demonstrate an immunoglobulin G inhibitor that selectively inhibits factor X activation. [117]
Treatment consists of plasma-derived factor X concentrate, prothrombin complex concentrate, or a plasma product such as FFP. [118] Treatment of acquired factor X deficiency is difficult. In 2001, Boggio and Green reported that control of bleeding with plasma or prothrombin complex concentrates is not completely successful. [119] Smith and colleagues had similar problems in 2 patients, which led them to resort to daily therapeutic plasma exchange with concomitant administration of intravenous immunoglobulin and steroids. [117] This therapy produced a rapid increase in factor X levels, which controlled the bleeding, followed by gradual recovery of normal factor X levels and correction of coagulation times. Splenectomy eliminates the acquired factor X deficiency in amyloidosis, but control of operative bleeding may require rFVIIa.
Factor XI deficiency
Factor XI deficiency is a congenital deficiency of blood coagulation factor XI (known as plasma thromboplastin antecedent [PTA] or antihemophilic factor C) resulting in a systemic blood-clotting defect called hemophilia C or Rosenthal syndrome, which may resemble classic hemophilia.
Factor XI is a key component of the intrinsic pathway of blood coagulation in vitro, but its exact role in vivo is uncertain. Factor XI is activated by thrombin and may participate in clot formation once coagulation has been initiated by other mechanisms. Additional coexisting abnormalities of hemostasis, such as von Willebrand disease, may also be responsible for variations in clinical presentation, particularly in individuals with mild factor XI deficiency. [120, 121]
Approximately 40-50% of all persons lacking factor XI are of Ashkenazi Jewish extraction. [122] Factor XI deficiency may be considered in patients evaluated for hemorrhage or unexplained, prolonged aPTT or through family or other genetic studies. Women with factor XI deficiency are prone to menorrhagia and to bleeding complications after childbirth. [123, 124] Individuals with factor XI deficiency need careful planning for elective surgery and dental extractions. FFP, fibrin glue, antifibrinolytic drugs, desmopressin, and factor XI concentrates have all been used successfully. Factor XI concentrate is usually reserved for younger patients with severe deficiency because its use in older patients has been associated with thrombotic phenomena.
Factor XII deficiency
Factor XII deficiency is defined as an absence or reduced level of blood coagulation factor XII (Hageman factor). Factor XII initiates the intrinsic coagulation cascade and is linked to the fibrinolytic, kallikrein-kinin, and complement systems. [125] It promotes the conversion of factor XI to its activated form. Factor XII deficiency typically occurs in the absence of a patient or family history of hemorrhagic disorders and is marked by prolonged clotting time.
Halbmayer et al have estimated the prevalence of severe and mild factor XII deficiency to be 1.5-3%. [126] This group has identified an association between factor XII deficiency and coronary artery disease. Measurements of plasma factor XII activity, fibrinogen, and lipoprotein in 426 consecutive patients with coronary heart disease awaiting cardiac surgery found 44 (10.3%) were moderately deficient in factor XII (factor XII activity, 17-50%; antigen, 15-57%). The prevalence of factor XII deficiency was significantly higher (P< 0.0001) among patients with coronary heart disease than among 300 similarly evaluated healthy blood donors (2.3%).
Factor XII deficiency has not been linked to any significant hemorrhagic diatheses. The disorder may be considered in patients with prolonged aPTT, normal PT, normal bleeding time, and no clinical history of bleeding. Once thought likely, the deficiency can be confirmed by normalization of aPTT with plasma component therapy and by factor assay.
Factor XII deficiency has clinical significance when attempts are made to heparinize individuals who have this condition. Routine coagulation tests used during cardiopulmonary bypass return abnormal findings in patients with factor XII deficiency and are useless for monitoring anticoagulation in these patients. Alternative monitoring systems, such as chromogenic heparin assay, citrated thrombin time, and recalcified thrombin time, must instead be used. [127]
Factor XIII deficiency
Factor XIII deficiency is a decrease or absence of factor XIII (fibrin-stabilizing factor [FSF]) that prevents blood-clot formation and results in a clinical hemorrhagic diathesis. Factor XIII is an enzyme found in plasma, platelets, and monocytes. In plasma, factor XIII has 2 subunits: the a subunit, which is the active enzyme and the b subunit, which is a carrier protein. [128] Activated factor XIII stimulates cross-linkage of fibrin as a means of stabilizing clot.
Bleeding in a patient with both normal PT and aPTT should raise the suspicion.
Congenital factor XIII deficiency is a severe autosomal recessive bleeding disorder associated with a characteristic pattern of neonatal hemorrhage and lifelong bleeding diathesis. Untreated patients have a high mortality rate. Even relatively minor trauma can be followed by prolonged and recurrent bleeding. Intracranial hemorrhage is a frequent complication. [129] The disorder affects both sexes, and bleeding may occur during pregnancy. [130] Acquired factor XIII deficiency has been described in HSP, various forms of colitis, erosive gastritis, and some forms of leukemia. Inhibitors to factor XIII are rare. [131]
Treatment of factor XIII deficiency requires lifelong prophylactic therapy with at least monthly infusions of factor XIII concentrate, even during pregnancy. [129, 130] . A recombinant factor XIII A-subunit (Tretten) is approved for routine prophylaxis of bleeding in patients with congenital factor XIII. [132]
Hemophilia A and B
The hemophilias are X-linked bleeding disorders caused by deficiency of factor VIII for hemophilia A and deficiency of factor IX for hemophilia B. The prevalence of hemophilia A is reported to be 17.1 per 100,000 males and for hemophilia B the prevalence is 3.8 per 100,000 males. [133]
Patients can have frequent, spontaneous, severe and even life-threatening bleeding including hemarthrosis, epistaxis, GI bleeding, and rarely intracranial bleeding. Severe hemophilia usually corresponds to factor activity levels of less than 1%. Female carriers may also manifest bleeding occurrences, especially in those with factor levels in the range of mild hemophilia. aPTT is prolonged, although in milder disease it may be normal. Diagnosis is made with confirmation of factor activity levels below 40% of normal. [134]
Treatment is primarily addressed with factor replacement; prophylactic factor replacement is standard practice for patients with severe disease. [118] For mucosal bleeding, antifibrinolytic therapy can be given as single agents or in combination with factor concentrates. They are not used in combination with aPCC due to increased risk of thrombosis. For mild hemophilia A, desmopressin may also be used to increase factor levels.
Factor VIII can be replaced with plasma-derived or recombinant factor concentrates, with newer products having a longer half-life for FVIII but no clinical superiority to older ones. The monoclonal antibody emicizumab, which performs the function of activated FVIII by bridging activated FIX and FX to restore the coagulation cascade, is approved for routine prophylaxis of bleeding episodes in patients with hemophilia A who have FVIII inhibitors. [135]
Recombinant factor IX concentrates are also available with extended half-ife factor IX concentrates having prolonged circulation times. Research efforts are also concentrated on gene therapy as an option for patients who require frequent factor concentrate infusions. In November 2022, the US Food and Drug Administration approved etranacogene dezaparvovec, as gene therapy for adults with hemophilia B. [134]
Questions & Answers
Overview
What are nonplatelet hemostatic disorders?
What are the coagulation-promoting conditions in nonplatelet hemostatic disorders?
What are the coagulation-impeding conditions in nonplatelet hemostatic disorders?
What are fibrinogen disorders?
What causes fibrinogen disorders?
What is the prevalence of congenital fibrinogen disorders?
Which clinical history findings are characteristic of fibrinogen disorders?
How are fibrinogen disorders diagnosed?
How are fibrinogen disorders treated?
What are the nonplatelet vascular and nonvascular hemostatic disorders?
What is the role of hyperglobulinemic purpura in the etiology of nonplatelet hemostatic disorders?
What is Henoch-Schönlein purpura (HSP)?
What is Waterhouse-Friderichsen syndrome?
What is Wiskott-Aldrich syndrome?
What is thrombotic thrombocytopenic purpura (TTP)?
Which inherited connective-tissue abnormalities cause vascular hemostatic disorders?
What are cavernous hemangiomata?
What is hereditary hemorrhagic telangiectasia?
What is the role of antithrombin III in the etiology of nonplatelet hemostatic disorders?
What is the role of protein C and S in the etiology of nonplatelet hemostatic disorders?
How is clinical thrombophilia defined in nonplatelet hemostatic disorders?
How is inherited thrombophilia diagnosed?
What is the risk of developing thrombosis in nonplatelet hemostatic disorders?
What are the racial predilections of nonplatelet hemostatic disorders?
How are coagulation-promoting nonplatelet hemostatic disorders diagnosed?
How are the factor V deficiencies differentiated and treated?
When are screening tests indicated for coagulation-promoting nonplatelet hemostatic disorders?
What is disseminated intravascular coagulation (DIC)?
What causes disseminated intravascular coagulation (DIC)?
What is the prevalence of disseminated intravascular coagulation (DIC)?
How is disseminated intravascular coagulation (DIC) diagnosed?
How is disseminated intravascular coagulation (DIC) treated?
What is factor VII deficiency?
What is factor XII deficiency?
What is factor XIII deficiency?
-
Approach to classification of nonplatelet hemostatic disorders.