Practice Essentials
Hemophilia A is an X-linked, recessive disorder caused by deficiency of functional plasma clotting factor VIII (FVIII), which may be inherited or arise from spontaneous mutation. The development of inhibitory alloantibodies to FVIII can severely complicate the treatment of genetic cases. Rarely, development of autoantibodies to FVIII results in acquired hemophilia A.
Signs and symptoms
Depending on the level of FVIII activity, patients with hemophilia may present with easy bruising; inadequate clotting of traumatic or even mild injury; or, in the case of severe hemophilia, spontaneous hemorrhage.
Signs of hemorrhage include the following:
-
General (usuall attributed to anemia secondary to bleeding): Weakness, orthostasis, tachycardia, tachypnea
-
Musculoskeletal (joints): Tingling, cracking, warmth, pain, stiffness, and refusal to use joint (children)
-
CNS: Headache, stiff neck, vomiting, lethargy, irritability, and spinal cord syndromes
-
Gastrointestinal: Hematemesis, melena, frank red blood per rectum, and abdominal pain
-
Genitourinary: Hematuria, renal colic, and post-circumcision bleeding
-
Other: Epistaxis, oral mucosal hemorrhage, hemoptysis, dyspnea (hematoma leading to airway obstruction), compartment syndrome symptoms, and contusions; excessive/prolonged bleeding with routine dental or other procedures
See Presentation for more detail.
Diagnosis
Laboratory studies for suspected hemophilia include the following:
-
Complete blood cell count
-
Screening coagulation studies (prothrombin time [PT], activated partial thromboplastin time [aPTT])
-
FVIII assay (clot based or chromogenic)
-
FVIII inhibitor assay (Bethesda assay, Nijmegen modified Bethesda assay)
Expected laboratory values are as follows:
-
Hemoglobin/hematocrit: Normal (or low if associated bleeding)
-
Platelet count: Normal
-
Prothrombin time (PT): Normal
-
APTT: Significantly prolonged in severe hemophilia, but may be normal or minimally prolonged in mild or even moderate hemophilia
Normal values for FVIII assays are 50-150%. Values in hemophilia A are as follows:
-
Mild: > 5%
-
Moderate: 1-5%
-
Severe: < 1%
Imaging studies for acute bleeds are chosen on the basis of clinical suspicion and anatomic location of involvement, as follows:
-
Head computed tomography scans without contrast are used to assess for spontaneous or traumatic intracranial hemorrhage
-
MRI scans of the head and spinal column are used for further assessment of spontaneous or traumatic hemorrhage
-
MRI is also useful in the evaluation of the cartilage, synovium, and joint space
-
Ultrasonography is useful in the evaluation of joints affected by acute or chronic effusions
Testing for inhibitors is indicated when bleeding is not controlled after infusion of adequate amounts of factor concentrate during a bleeding episode. The presence of inhibitors is indicated by failure of correction of clotting times with 1:1 mix with normal plasma. Inhibitor concentration is titrated using the Bethesda method, as follows:
-
Positive result: ≥ 0.6 Bethesda units (BU)
-
Low-titer inhibitor: ≤5 BU
-
High-titer inhibitor: >5 BU
See Workup for more detail.
Management
The treatment of hemophilia may involve the following:
-
Management of hemostasis
-
Management of bleeding episodes including hemostatic support and pain management
-
Use of factor replacement products and adjuvant medications
-
Gene therapy
-
Treatment of patients with factor inhibitors
-
Treatment and rehabilitation of patients with hemophilia synovitis
Disposition of treatment is as follows:
-
Management ideally should be provided through a comprehensive hemophilia care center
-
Home administration of treatment and infusions by the family or patient is customary
-
FVIII treatment may be given prophylactically or on demand
-
Hospitalization is reserved for severe or life-threatening bleeds or for patients for whom home infusion is unavailable or impractical
For treatment of acute bleeds, target levels by hemorrhage severity are as follows:
-
Mild hemorrhages (eg, early hemarthrosis, epistaxis, gingival bleeding): Maintain an FVIII level of 30%
-
Major hemorrhages (eg, late hemarthrosis, muscle bleeds): Maintain an FVIII level of at least 50%
-
Life- or limb-threatening bleeding episodes (eg, major trauma or surgery, advanced or recurrent hemarthrosis, major GI bleeding, any head trauma, signs of distal neurovascular compormise of limb or compartment syndrome): Maintain an FVIII level of 80-100%
Ideally, therapy is individualized to specific patients. However, for general dosing, to find the number of units of factor VIII needed to correct the factor VIII activity level, use the following formula:
Dose in FVIII IU = (weight in kg) x (desired FVIII increase) x (0.5 IU/kg per IU/dL)
FVIII regimens are as follows:
-
The second dose should be administered 8-12 hours after the initial dose and is usually one half the initial calculated dose
-
Minor hemorrhage requires 1-3 doses of FVIII
-
Major hemorrhage requires many doses and continued FVIII activity monitoring with the goal of keeping the trough activity level at least 50%
-
Continuous infusions of FVIII may be considered for major hemorrhage or major surgery
The following types of FVIII concentrates are available:
-
Plasma-based products: Purified to inactivate viruses
-
First-generation recombinant products: Produced in mammalian cell lines, contain animal and/or human plasma-derived proteins in cell culture media and in final product.
-
Second-generation recombinant products: Produced in mammalian cell lines, contain animal and/or human plasma-derived proteins in cell culture media but none in final product.
-
Third-generation recombinant products: Produced in mammalian cell lines, contain no animal and/or human plasma-derived proteins in cell culture media or in final product.
-
Extended half-life recombinant FVIII products
Desmopressin vasopressin analog, or 1-deamino-8-D-arginine vasopressin (DDAVP), has the following attributes:
-
Depending on baseline level and goal factor level, may be considered the treatment of choice for mild and moderate hemophilia A
-
Not effective in the treatment of severe hemophilia
-
Can be intravenously administered at a dose of 0.3 mcg/kg of body weight in the inpatient setting (Canadian labeling recommends maximum dose of 20 mcg).
-
Peak effect is observed in 30-60 minutes
-
DDAVP leads to free water retention, which can lead to hyponatremia
-
A concentrated DDAVP intranasal spray is available for outpatient use
The following antifibrinolytics are used in addition to FVIII replacement for oral mucosal hemorrhage and prophylaxis:
-
Epsilon aminocaproic acid (Amicar)
-
Tranexamic acid (Cyklokapron, Lyseda)
Treatments used in patients with inhibitors of FVIII are as follows:
-
High doses of FVIII for low-titer inhibitors
-
Activated prothrombin complex concentrate (aPCC)
-
Activated recombinant FVII (rFVIIa)
-
Monoclonal antibodies directed toward restoring FVIII function (eg, emicizumab)
-
Porcine FVIII, which has low cross-reactivity with human FVIII antibody
-
Desensitization
-
Immune tolerance induction (ITI)
See Treatment and Medication for more detail.
COVID-19
The World Federation of Hemophilia (WFH) advises that no increased susceptibility to SARS-CoV-2 infection has been found in immunocompetent patients with bleeding disorders, and there is currently no known COVID-19 risk from blood, blood treatment products, and plasma-derived products. [1]
In addition, the WFH notes the following:
-
As COVID-19 progresses, coagulation pathways are activated as part of the host inflammatory response to limit the viral infection. Specifically, D-dimer levels are often elevated. More severe COVID-19 may lead to overt disseminated intravascular coagulation (DIC), associated with high mortality. Close monitoring for bleeding and thrombosis is recommended for all individuals who progress with signs or symptoms of DIC.
-
Anticoagulants (eg, low molecular weight heparin [LMWH]) are being recommended as part of treatment protocols for patients with elevated D-dimers and severe infection. Use of anticoagulants should be accompanied by factor replacement therapy.
-
If COVID-19 is diagnosed, prophylaxis with factor replacement therapy should be continued, with consideration of higher trough levels (like those for major trauma) in patients hospitalized for severe infection.
-
The risk of COVID-19–related thrombotic complications for hemophilia patients receiving treatment with non–factor replacement therapies, such emicizumab or other investigational agents (eg, fitusiran, anti–tissue factor pathway inhibitor) is unknown.
-
Whether emicizumab may interact with COVID-19–related coagulopathy is unknown; close monitoring for thrombosis is recommended. Prophylaxis should be continued, and in the event of missed doses, the long half-life (~30 days) of emicizumab should be taken into consideration, since it will be present and active for a prolonged period of time.
-
Patients should be assessed to determine whether they need additional clotting factor replacement therapy.
-
Anticoagulants may be considered as per recommended treatment protocols.
-
In patients with FVIII inhibitors receiving emicizumab, extra precautions should be taken if they require aPCC, because of the known drug-drug interaction between emicizumab and aPCC.
-
Some one-stage coagulation assays—such as aPTT, which is often used to diagnose and monitor patients in DIC—overestimate coagulation in patients on emicizumab and thus may mask coagulopathy.
-
For patients who are participating in clinical studies, investigators are advised to seek guidance from the study sponsors and medical monitors. Patients should inform health care providers they are in a clinical study; consultation with their hematologist is recommended.
-
For patients participating in a clinical trial of gene therapy, in addition to caution regarding infection risk due to immunosuppression, supplementation to higher coagulation factor levels (eg, as if treating major trauma) could be considered in those who have had a suboptimal response to the gene therapy.
-
Patients with bleeding disorders of all severities and COVID-19 should be eligible for all available therapies that would be required depending on their condition (eg, ventilation support, extracorporeal membrane oxygenation [ECMO], hemofiltration). Having hemophilia should not exclude individuals from invasive management of COVID-19.
A case report of COVID-19 in a patient in Wuhan, China, suggests that home management with active monitoring is appropriate for patients with hemophilia who have mild cases of COVID-19. Such patients may benefit from administration of replacement factors at the onset of infectious illness. [2]
The WFH, in collaboration with the European Association for Haemophilia and Allied Disorders, European Haemophilia Consortium, and US National Hemophilia Foundation, has also issued recommendations on COVID-19 vaccination for people with bleeding disorders. [3]
Background
Hemophilia A is an inherited, X-linked, recessive disorder caused by deficiency of functional plasma clotting factor VIII (FVIII). In a significant number of cases, the disorder results from a new mutation. Rarely this develops as an acquired auto-immune process.
Morbidity and death are primarily the result of hemorrhage, although infectious diseases (eg, HIV infection, hepatitis) became prominent, particularly in patients who received blood products prior to 1985.
Laboratory studies for suspected hemophilia include a complete blood cell count, coagulation studies, and an FVIII assay. In patients with an established diagnosis of hemophilia, periodic laboratory evaluations include screening for the presence of FVIII inhibitor and screening for transfusion-related or transmissible diseases such as hepatitis and HIV infection. Measurement of FVIII levels is important for monitoring FVIII replacement therapy. (See Workup.)
The treatment of hemophilia may involve prophylaxis, management of bleeding episodes, immune tolerance induction for patients with factor inhibitors, and treatment and rehabilitation of patients with hemophilia synovitis. Treatment of patients with hemophilia ideally should be provided through a comprehensive hemophilia care center (see Treatment and Medication). [4]
Please see the following for more information:
Classification
The classification of the severity of hemophilia has been based on either clinical bleeding symptoms or on plasma procoagulant levels; the latter are the most widely used criteria. Classification according to plasma procoagulant levels is as follows:
-
Severe hemophilia - FVIII level less than 1% of normal (< 0.01 IU/mL)
-
Moderate hemophilia - FVIII level 1-5% of normal (0.01-0.05 IU/mL)
-
Mild hemophilia - FVIII level more than 5% but less than 40% of normal (>0.05 to < 0.40 IU/mL)
Severe disease presents in children younger than 1 year and accounts for 43-70% of hemophilia A cases. Moderate disease presents in children aged 1-2 years and accounts for 15-26% of cases. Mild disease presents in children older than 2 years and accounts for 15-31% of cases.
Clinical bleeding symptom criteria have been used because patients with FVIII levels of less than 1% occasionally have little or no spontaneous bleeding and appear to have clinically moderate or mild hemophilia. Furthermore, the reverse is true for patients with procoagulant activities of 1-5%, who may present with clinically severe disease.
Historical background
Hemophilia is one of the oldest described genetic diseases. An inherited bleeding disorder in males was recognized in Talmudic records of the second century.
The modern history of hemophilia began in 1803 with the description of hemophilic kindred by John Otto, followed by the first review of hemophilia by Nasse in 1820. Wright demonstrated evidence of laboratory defects in blood clotting in 1893; however, FVIII was not identified until 1937, when Patek and Taylor isolated a clotting factor from the blood, which they called antihemophilic factor (AHF).
A bioassay of FVIII was introduced in 1950. Although the intimate relationship between FVIII and von Willebrand factor (vWF) is now known, it was not appreciated at the time. In 1953, decreased FVIII levels in patients with vWF deficiency was first described. Further research by Nilson and coworkers indicated the interaction between these 2 clotting factors.
In 1952, hemophilia B was described and was named Christmas disease after the surname of the first patient who was examined in detail. The differentiation of hemophilia B from hemophilia A followed the observation that mixing plasma from a patient with "true hemophilia" with plasma from a patient with Christmas disease corrected the clotting time. Hemophilia A makes up approximately 80% of hemophilia cases.
In the early 1960s, cryoprecipitate (the precipitate from fresh frozen plasma that has been thawed and centrifuged) became the first concentrate available for the treatment of patients with hemophilia. In the 1970s, lyophilized (ie, freeze-dried) intermediate-purity concentrates were obtained from large pools of blood donors. The introduction of concentrated lyophilized products that are easy to store and transport dramatically improved the quality of life of patients with hemophilia and facilitated their preparation for surgery and home care.
Unfortunately, the large size of the donor pool—as many as 20,000 donors may contribute to a single lot of plasma-derived FVIII concentrate—heightened the risk of viral contamination of commercial FVIII concentrates. By the mid-1980s, the majority of patients with severe hemophilia had been exposed to hepatitis A, hepatitis B, and hepatitis C viruses and human immunodeficiency virus (HIV).
Viricidal treatment of plasma-derived FVIII concentrates has been effective in eliminating new HIV transmissions and virtually eliminating hepatitis B and hepatitis C exposures. The introduction of recombinant FVIII concentrate, and the gradual elimination of albumin from the production process used for these products, has virtually eliminated the risk of viral exposure.
Pathophysiology
Factor VIII production, processing, and structure
Primary sites of factor VIII (FVIII) production are thought to be the vascular endothelium in the liver and the reticuloendothelial system. Liver transplantation corrects FVIII deficiency in persons with hemophilia.
FVIII messenger RNA has been detected in the liver, spleen, and other tissues. [5] Studies of FVIII production in transfected cell lines have shown that following synthesis, FVIII moves to the lumen of the endoplasmic reticulum, where it is bound to several proteins that regulate secretion, particularly immunoglobulin binding protein, from which it has to dissociate in an energy-dependent process.
Cleavage of FVIII's signal peptide and the addition of oligosaccharides also occur in the endoplasmic reticulum. The chaperone proteins, calnexin and calreticulin, enhance both FVIII secretion and degradation.
A part of the factor FVIII protein in the endoplasmic reticulum is degraded within the cell. The other part enters the Golgi apparatus, where several changes occur to produce the heavy and light chains and to modify the carbohydrates. The addition of sulfates to tyrosine residues of the heavy and light chains is necessary for full procoagulant activity, with the sulfated region playing a role in thrombin interaction. This posttranslational sulfation of tyrosine residues impacts the procoagulant activity of factor VIII and its interaction with von Willebrand factor (vWF).
von Willebrand factor
FVIII circulates in plasma in a noncovalently bound complex with vWF, which plays significant roles in the function, production, stabilization, conformation, and immunogenicity of FVIII. [6] VWF has been termed FVIII-related antigen (FVIII-R); related terminology for FVIII is FVIII-coagulant (FVIII-C).
VWF appears to promote assembly of the heavy and light chains of FVIII and more efficient secretion of FVIII from the endoplasmic reticulum. It also directs FVIII into the Weibel-Palade bodies, which are the intracellular storage sites for vWF.
In plasma, vWF stabilizes FVIII and protects it from degradation. In the presence of normal vWF protein, the half-life of FVIII is approximately 12 hours, whereas in the absence of vWF, the half-life of FVIII-C is reduced to 2 hours. [7, 8, 9]
The clotting cascade
The role of the coagulation system is to produce a stable fibrin clot at sites of injury. The clotting mechanism has two pathways: intrinsic and extrinsic. See the image below.
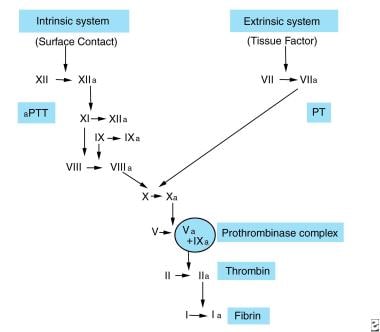
The intrinsic system is initiated when factor XII is activated by contact with damaged endothelium. The activation of factor XII can also initiate the extrinsic pathway, fibrinolysis, kinin generation, and complement activation.
In conjunction with high-molecular-weight kininogen (HMWK), factor XIIa converts prekallikrein (PK) to kallikrein and activates factor XI. Activated factor XI, in turn, activates factor IX in a calcium-dependent reaction. Factor IXa can bind phospholipids. Then, factor X is activated on the phospholipid surface; activation of factor X involves a complex (tenase complex) of factor IXa, thrombin-activated FVIII, calcium ions, and phospholipid.
In the extrinsic system, the conversion of factor X to factor Xa involves tissue factor (TF), or thromboplastin; factor VII; and calcium ions. TF is released from the damaged cells and is thought to be a lipoprotein complex that acts as a cell surface receptor for factor VII, with its resultant activation. TF also adsorbs factor X to enhance the reaction between factor VIIa, factor X, and calcium ions. Factor IXa and factor XII fragments can also activate factor VII.
In the common pathway, factor Xa (generated through the intrinsic or extrinsic pathways) forms a prothrombinase complex with phospholipids, calcium ions, and thrombin-activated factor Va. The complex cleaves prothrombin into thrombin and prothrombin fragments 1 and 2.
Thrombin converts fibrinogen into fibrin and activates FVIII, factor V, and factor XIII. Fibrinopeptides A and B, the results of the cleavage of peptides A and B by thrombin, cause fibrin monomers to form and then polymerize into a meshwork of fibrin; the resultant clot is stabilized by factor XIIIa and the cross-linking of adjacent fibrin strands.
Because of the complex interactions of the intrinsic and extrinsic pathways (factor IXa activates factor VII), the existence of only one in vivo pathway with different mechanisms of activation has been suggested. See the image below.
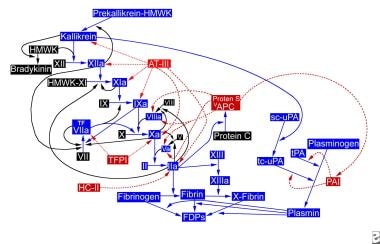 The hemostatic pathway. APC = activated protein C (APC); AT-III = antithrombin III; FDP = fibrin degradation products; HC-II = heparin cofactor II; HMWK = high-molecular-weight kininogen; PAI = plasminogen activator inhibitor; sc-uPA = single-chain urokinase plasminogen activator; tc-uPA = two-chain urokinase plasminogen activator; TFPI = tissue factor pathway inhibitor; tPA = tissue plasminogen activator
The hemostatic pathway. APC = activated protein C (APC); AT-III = antithrombin III; FDP = fibrin degradation products; HC-II = heparin cofactor II; HMWK = high-molecular-weight kininogen; PAI = plasminogen activator inhibitor; sc-uPA = single-chain urokinase plasminogen activator; tc-uPA = two-chain urokinase plasminogen activator; TFPI = tissue factor pathway inhibitor; tPA = tissue plasminogen activator
FVIII and factor IX circulate in an inactive form. When activated, these 2 factors cooperate to cleave and activate factor X, a key enzyme that controls the conversion of fibrinogen to fibrin. Therefore, the lack of FVIII may significantly alter clot formation and, as a consequence, result in clinical bleeding.
Genetics
The gene for FVIII (F8C) is located on the long arm of chromosome X, within the Xq28 region. The gene is unusually large, representing 186 kb of the X chromosome. It comprises 26 exons and 25 introns. Mature FVIII contains 2332 amino acids. See the image below.
 Structural domains of human factor VIII. Adapted from: Stoilova-McPhie S, Villoutreix BO, Mertens K, Kemball-Cook G, Holzenburg A. 3-Dimensional structure of membrane-bound coagulation factor VIII: modeling of the factor VIII heterodimer within a 3-dimensional density map derived by electron crystallography. Blood. Feb 15 2002;99(4):1215-23; Roberts HR, Hoffman M. Hemophilia A and B. In: Beutler E, Lichtman MA, Coller BS, et al, eds. Williams Hematology. 6th ed. NY: McGraw-Hill; 2001:1639-57; and Roberts HR. Thoughts on the mechanism of action of FVIIa. Presented at: Second Symposium on New Aspects of Haemophilia Treatment; 1991; Copenhagen, Denmark.
Structural domains of human factor VIII. Adapted from: Stoilova-McPhie S, Villoutreix BO, Mertens K, Kemball-Cook G, Holzenburg A. 3-Dimensional structure of membrane-bound coagulation factor VIII: modeling of the factor VIII heterodimer within a 3-dimensional density map derived by electron crystallography. Blood. Feb 15 2002;99(4):1215-23; Roberts HR, Hoffman M. Hemophilia A and B. In: Beutler E, Lichtman MA, Coller BS, et al, eds. Williams Hematology. 6th ed. NY: McGraw-Hill; 2001:1639-57; and Roberts HR. Thoughts on the mechanism of action of FVIIa. Presented at: Second Symposium on New Aspects of Haemophilia Treatment; 1991; Copenhagen, Denmark.
Approximately 40% of cases of severe FVIII deficiency arise from a large inversion that disrupts the FVIII gene. Deletions, insertions, and point mutations account for the remaining 50-60% of the F8C defects that cause hemophilia A.
Low FVIII levels may arise from defects outside the FVIII gene, as in type IIN von Willebrand disease, in which the molecular defect resides in the FVIII-binding domain of von Willebrand factor.
Hemophilia A
FVIII deficiency, dysfunctional FVIII, or FVIII inhibitors lead to the disruption of the normal intrinsic coagulation cascade, resulting in excessive hemorrhage in response to trauma and, in severe cases, spontaneous hemorrhage. Hemorrhage sites include joints (eg, knee, elbow); muscles; the central nervous system (CNS); and the gastrointestinal, genitourinary, pulmonary, and cardiovascular systems. Intracranial hemorrhage occurs most commonly in patients younger than 18 years and can be fatal.
Hemorrhage into joints
The hallmark of hemophilia is hemorrhage into joints. This bleeding is painful and leads to long-term inflammation and deterioration of the joint.
Human synovial cells synthesize high levels of tissue factor pathway inhibitor, resulting in a higher degree of factor Xa (FXa) inhibition, which predisposes hemophilic joints to bleed. This effect may also account for the dramatic response of activated factor VII (FVIIa) infusions in patients with acute hemarthroses and FVIII inhibitors.
Bleeding into a joint may lead to synovial inflammation, which predisposes the joint to further bleeds. A joint that has had repeated bleeds (by one definition, at least 4 bleeds within a 6-month period) is termed a target joint. Commonly, this occurs in knees though ankles and elbows are other commonly affected joints.
Repeated hemarthroses lead to progressive synovial hypertrophy, hemosiderin deposition, fibrosis, and damage to cartilage, with subchondral bone-cyst formation. This results in permanent deformities, misalignment, loss of mobility, and extremities of unequal lengths.
Inhibitors
Approximately 30% of patients with severe hemophilia A develop alloantibody inhibitors that can bind FVIII. These inhibitors are typically immunoglobulin G (IgG), predominantly of the IgG4 subclass, that neutralize the coagulant effects of replacement therapy. However, the inhibitors do not fix complement and do not result in the end-organ damage observed with circulating immune complexes
Inhibitors occur at a young age (about 50% by age 10 years), principally in patients with less than 1% FVIII. Both genetic and environmental factors determine the frequency of inhibitor development. Specific molecular abnormalities (eg, gene deletions, stop codon mutations, frameshift mutations) are associated with a higher incidence of inhibitor development. In addition, inhibitors are more likely to develop in black children. Missense mutations are associated with a low risk of inhibitor development. [10]
The association of product used with the risk of inhibitor formation remains controversial. In a study of 574 patients with severe hemophilia A, 177 of whom developed inhibitors, the risks of inhibitor development were similar with recombinant and plasma-derived FVIII products. No association was found between the development of inhibitors and the von Willebrand factor content of products, switching from a plasma-derived to a recombinant product, or switching among brands of FVIII products. Unexpectedly, however, inhibitors developed more often with second-generation full-length recombinant products than with third-generation products. [11]
A study with 303 previously untreated or minimally treated children with hemophilia demonstrated a increased risk of inhibitor formation in patients who used recombinant products. [12] However, the European Medicines Agency (EMA) Pharmacovigilance Risk Assessment Committee (PRAC) concluded that "there is no clear and consistent evidence of a difference in the incidence of inhibitor development between the two classes of factor VIII medicines: those derived from plasma and those made by recombinant DNA technology." [13]
Due to conflicting data and challenging methodological limitations, choice of initial treatment in previously untreated or minimally treated patients should be done in a colaborative fashion between patients/caregivers and treatment teams wtih experience in these issues.
In the United States, levels of FVIII inhibitors are most often measured by the Bethesda method. In this method, 1 Bethesda unit (BU) equals the amount of antibody that destroys one half of the FVIII in an equal mixture of normal plasma and patient plasma in 2 hours at 37°C. Inhibitor levels are described as low titer or high titer, depending on whether they are less than or more than 5 BU, respectively; high-titer inhibitor levels are typically far higher than 5 BU.
The Nijmegen modification uses immunodepleted FVIII–deficient plasma instead of an imidazole saline buffer to ensure pH control to prevent non–antibody-mediated loss of FVIII-C activity during the prolonged 2-hour incubation period. [14] The Bethesda assay tends to underestimate the titer of VIII autoantibody because of its characteristics, in contrast to a hemophilic antibody. [15] The Oxford assay is another modification of the Bethesda inhibitor test.
Acquired hemophilia
Acquired hemophilia is the development of FVIII inhibitors (autoantibodies) in persons without a history of FVIII deficiency. This condition can be idiopathic (occurring usually in people >50 y). It can be associated with underlying collagen vascular disease or the peripartum period, or it may represent a drug reaction (eg, to penicillin). High titers of FVIII autoantibodies may be associated with malignancies, particularly lymphoproliferative malignancies. [16]
Etiology
Hemophilia A is caused by an inherited or acquired genetic mutation that results in dysfunction or deficiency of factor VIII, or by an acquired inhibitor that binds factor VIII. Of genetic cases, up to approximately one third are the result of de novo mutations not present in the mother's X chromosome.
Inadequate factor VIII results in the insufficient generation of thrombin by the FIXa and FVIIIa complex by means of the intrinsic pathway of the coagulation cascade. This mechanism, in combination with the effect of the tissue-factor pathway inhibitor, creates an extraordinary tendency for impaired clotting in response to trauma and, especially in persons with severe hemophilia, with spontaneous bleeding.
Hemophilia A is inherited in an X-linked recessive pattern. The gene for FVIII is located on the long arm of the X chromosome in band q28. The factor VIII gene is one of the largest genes, comprising approximately 0.1% of the DNA in the X chromosome; it is 186 kilobases (kb) long and has a 9-kb coding region that contains 26 exons. The mature protein contains 2332 amino acids and has a molecular weight of 300 kd. It includes 3 A domains, 1 B domain, and 2 C domains.
Intron 22 of the factor VIII gene, uniquely, contains two other genes. The first gene, F8A, is transcribed in a direction opposite to that of the factor VIII gene itself. The second gene, F8B, is transcribed in the 3' (normal) direction similar to the factor VIII gene. Sequences called A2 and A3, homologous to the F8A sequence, are present on the X chromosome, 300 kb telomeric to the factor VIII gene.
Homologous recombination of the factor VIII gene, with inversion and crossover involving the F8A sequence in intron 22 and the homologous distal sequence on the X chromosome, results in a split in the factor VIII gene with the two parts aligned in opposite directions. This causes a disruption in the normal factor VIII coding sequence, with an inability to transcribe the complete, normal factor VIII protein, resulting in a loss of function.
The mutation in intron 22 occurs during spermatogenesis and is a common cause of severe factor VIII deficiency; it is present in approximately 40% of patients. It is easily detected using a Southern blot analysis of the patient's DNA. These patients are more likely to develop an inhibitor to factor VIII.
In one study, all detected inversions originated in a maternal grandparent during male meiosis (spermatogenesis), supporting the hypothesis that an unpaired Xq, rather than a paired X chromosome, is more likely to undergo an intrachromosomal inversion. The majority of mothers of persons with the sporadic, inversion-related severe hemophilia are carriers. [17]
The knowledge of the parental origin of the inversion mutation has important implications for genetic counseling.
Several other types of mutations have been described. Point mutations can lead to mild, moderate, or severe deficiency of factor VIII, depending on the effect of that mutation on factor VIII gene function.
Missense mutations, such as the G-to-A single-base substitution, alter the amino acid composition of the molecule, producing a dysfunctional molecule (FVIII antigen present with reduction in FVIII activity); these mutations are associated with mild, moderate, or severe factor VIII reductions and are associated with the development of factor VIII inhibitors. Intracellular accumulation of factor VIII induced by Arg 593→Cys and Asn 618→Ser missense mutations also result in reduction of cross-reacting material in severe hemophilia A.
Gene deletions lead to factor VIII deficiency, and large gene deletions result in severe hemophilia, with no detectable factor VIII antigen; such patients are more susceptible to inhibitor development. Insertions are apparently uncommon in the factor VIII gene, but they usually lead to severe hemophilia A. [18] Nonsense mutations and abnormal splicing may also occur.
Other causes of this disorder remain to be identified. The Haemophilia A Mutation, Structure, Test and Resource Site (HAMSTeRS) has a continually updated database of genetic defects related to hemophilia A.
Combined factor V and factor VIII deficiency
Combined FV and FVIII deficiency is an autosomal recessive disorder, with clinical manifestations in affected females and males. The disorder is caused by mutations in one of two genes, lectin mannose binding protein 1 (LMAN1) or multiple coagulation factor deficiency 2 (MCFD2), which encode proteins involved in the intracellular transport of FV and FVIII; the coagulation factors themselves are normal. [19]
Epidemiology
Hemophilia A is the most common X-linked genetic disease and the second most common factor deficiency after von Willebrand disease (vWD). The worldwide incidence of hemophilia A is approximately 1 case per 5000 males, with approximately one third of affected individuals not having a family history of the disorder. The prevalence of hemophilia A varies with the reporting country, with a range of 5.4-14.5 cases per 100,000 males. During the period 2012-2018, the number of males in the United States with hemophilia was estimated to be about 33,000. [20]
Approximately 50-60% of patients have severe hemophilia A (FVIII < 2% of normal), associated with the severest bleeding manifestations. Approximately 25-30% have moderate hemophilia (FVIII 2-5%) and manifest bleeding after minor trauma. Those with mild hemophilia A (FVIII 6-30%) comprise 15-20% of all people with hemophilia; these patients develop bleeding only after trauma or surgery.
Acquired hemophilia A, caused by the development of an autoantibody to FVIII in a person with previously normal hemostasis, develops with a frequency of 1 case per 1 million population per year. [21] Acquired FVIII deficiency is observed in older populations, generally those older than 60 years.
The inherited, combined deficiency of factors V and VIII is a rare but recognized cause of a bleeding disorder. The prevalence is estimated to be 1 case per million population. [19]
Racial, sexual, and age-related differences in incidence
Hemophilia A occurs in all races and ethnic groups. In general, the demographics of hemophilia follow the racial distribution in a given population; for example, rates of hemophilia among whites, African Americans, and Hispanics in the US are similar.
Because hemophilia is an X-linked, recessive condition, it occurs predominantly in males. Females usually are asymptomatic carriers. However, mild hemophilia may be more common in carriers than previously recognized. In 1 study, 5 of 55 patients with mild hemophilia (factor levels 5-50%) were girls. [22]
Females may have clinical bleeding due to hemophilia if any of the following 3 conditions is present:
-
Extreme lyonization (ie, inactivation of the normal FVIII allele in one of the X chromosomes) [23]
-
Homozygosity for the hemophilia gene (ie, father with hemophilia and mother who is a carrier, two independent mutations, or some combination of inheritance and new mutations)
-
Turner syndrome (XO) associated with the affected hemophilia gene
In genetic cases, significant deficiency in FVIII may be evident in the neonatal period. It continues through the life of the affected individual. The absence of hemorrhagic manifestations at birth does not exclude hemophilia.
Prognosis
With appropriate education and treatment, patients with hemophilia can live full and productive lives. Prophylaxis and early treatment with FVIII concentrate that is safe from viral contamination have dramatically improved the prognosis of patients with severe hemophilia. Nevertheless, approximately one quarter of patients with severe hemophilia aged 6-18 years have below-normal motor skills and academic performance and have more emotional and behavioral problems than others. [24]
Factor concentrates have made home-replacement therapy possible, improving patients' quality of life. In addition, the era of replacement therapy brought dramatic gains in life expectancy. For patients with severe hemophilia, life expectancy rose from 11 years or less before the 1960s to almost 60 years prior to HIV epidemic in the 1980s. [5, 7]
Increasing evidence associates hemophilia with low bone mineral density and increased fracture risk in both children and adults. Physical inactivity (which may be worsened by arthropathy) and vitamin D deficiency seem to play a fundamental role. [25]
Viral infection from contaminated FVIII concentrate became a problem during the replacement era. Most patients with hemophilia who received plasma-derived products that were not treated to eliminate potential contaminating viruses became infected with HIV or hepatitis A, hepatitis B, or hepatitis C viruses.
The most serious of these was HIV infection. The first deaths of people with hemophilia due to AIDS were observed in the early 1980s. Rates of seroconversion were more than 75% for those with severe disease, 46% for moderate disease, and 25% for mild disease.
In the United States, death rates of patients with hemophilia increased from 0.4 deaths per million population in 1979-1981 to 1.2 deaths per million population in 1987-1989; AIDS accounted for 55% of all hemophilia deaths. Causes of death shifted from intracranial and other bleeding to AIDS and cirrhosis from hepatitis. AIDS remains the most common cause of death in patients with severe hemophilia. [7] Indeed, HIV-infected individuals are more likely to die of that disease than from hemophilia.
With improved screening of donors, new methods of factor concentrate purification, and recombinant concentrates, infectious complications now are only historically important. However, even with these methods, some viruses (eg, parvovirus B-19) cannot be removed and may be transmitted through plasma-derived products. Other potential infectious agents include the prions that cause Creutzfeldt-Jakob disease. With the development of animal protein–free products, the risk of contamination with these agents may be decreased.
Intracranial hemorrhage and hemorrhages into the soft tissue around vital areas, such as the airway or internal organs, remain the most important life-threatening complications. The lifetime risk of intracranial bleeding is 2-8% and accounts for one third of deaths due to hemorrhage, even in the era of factor replacement. Intracranial hemorrhage is the second most common cause of death and the most common cause of death related to hemorrhage. Of patients with severe hemophilia, 10% have intracranial bleeding, with a mortality rate of 30%.
Chronic debilitating joint disease results from repeated hemarthrosis; synovial membrane inflammation; hypertrophy; and, eventually, destructive arthritis. Early replacement of coagulation factors by means of infusion is essential to prevent functional disability. Thus, prophylactic therapy administered 2-3 times weekly, starting when patients are young, is considered the standard of care in most developed countries.
Before the widespread use of replacement therapy, patients with severe hemophilia had a shortened lifespan and diminished quality of life that was greatly affected by hemophilic arthropathy. Home therapy for hemarthroses became possible with factor concentrates. Prophylactic use of lyophilized concentrates that eliminate bleeding episodes help prevent joint deterioration, especially when instituted early in life (ie, at age 1-2 y).
Overall, the mortality rate for patients with hemophilia is twice that of the healthy male population. For severe hemophilia, the rate is 4-6 times higher. If hepatitis and cirrhosis are excluded, the overall mortality rate of patients with severe hemophilia A is 1.2 times that of the healthy male population. [7]
Patient Education
Starting in infancy, regular dental evaluation is recommended, along with instruction regarding proper oral hygiene, dental care, and adequate fluoridation. Encourage the patient to engage in appropriate exercise. Advise the patient against participating in contact and collision sports.
Patient and family education about early recognition of hemorrhage signs and symptoms is important for instituting or increasing the intensity of replacement therapy. This treatment helps prevent the acute and chronic complications of the disease, which range from those that can impair quality of life to those that are life-threatening.
Educating patients and family members about factor replacement administration at home has greatly enhanced the quality of life of patients with severe hemophilia by allowing prompt infusion for bleeds and markedly reducing the need for emergency department visits. Parents often can learn to infuse children as young as 2 years, and, by 8-10 years, most children with hemophilia can learn to self-infuse.
For patient education information, see Hemophilia.
-
Coagulation pathway.
-
The hemostatic pathway. APC = activated protein C (APC); AT-III = antithrombin III; FDP = fibrin degradation products; HC-II = heparin cofactor II; HMWK = high-molecular-weight kininogen; PAI = plasminogen activator inhibitor; sc-uPA = single-chain urokinase plasminogen activator; tc-uPA = two-chain urokinase plasminogen activator; TFPI = tissue factor pathway inhibitor; tPA = tissue plasminogen activator
-
Structural domains of human factor VIII. Adapted from: Stoilova-McPhie S, Villoutreix BO, Mertens K, Kemball-Cook G, Holzenburg A. 3-Dimensional structure of membrane-bound coagulation factor VIII: modeling of the factor VIII heterodimer within a 3-dimensional density map derived by electron crystallography. Blood. Feb 15 2002;99(4):1215-23; Roberts HR, Hoffman M. Hemophilia A and B. In: Beutler E, Lichtman MA, Coller BS, et al, eds. Williams Hematology. 6th ed. NY: McGraw-Hill; 2001:1639-57; and Roberts HR. Thoughts on the mechanism of action of FVIIa. Presented at: Second Symposium on New Aspects of Haemophilia Treatment; 1991; Copenhagen, Denmark.
-
Possible genetic outcomes in individuals carrying the hemophilic gene.
-
Photograph of a teenage boy with bleeding into his right thigh as well as both knees and ankles.
-
Photograph of the right knee in an older man with a chronically fused, extended knee following open drainage of knee bleeding that occurred many years previously.
-
Photograph depicting severe bilateral hemophilic arthropathy and muscle wasting. The 3 punctures made into the left knee joint were performed in an attempt to aspirate recent aggravated bleeding.
-
Radiograph depicting advanced hemophilic arthropathy of the knee joint. These images show chronic severe arthritis, fusion, loss of cartilage, and joint space deformities.
-
Radiograph depicting advanced hemophilic arthropathy of the elbow. This image shows chronic severe arthritis, fusion, loss of cartilage, and joint space deformities.
-
Photograph of a hemophilic knee at surgery, with synovial proliferation caused by repeated bleeding; synovectomy was required.
-
Large amount of vascular synovium removed at surgery.
-
Microscopic appearance of synovial proliferation and high vascularity. If stained with iron, diffuse deposits would be demonstrated; iron-laden macrophages are present.
-
Large pseudocyst involving the left proximal femur.
-
Transected pseudocyst (following disarticulation of the left lower extremity due to vascular compromise, nerve damage, loss of bone, and nonfunctional limb). This photo shows black-brown old blood, residual muscle, and bone.
-
Dissection of a pseudocyst.
-
Transected pseudocyst with chocolate brown-black old blood.
-
Photograph of a patient who presented with a slowly expanding abdominal and flank mass, as well as increasing pain, inability to eat, weight loss, and weakness of his lower extremity.
-
Plain radiograph of the pelvis showing a large lytic area.
-
Intravenous pyelogram showing extreme displacement of the left kidney and ureter by a pseudocyst.
-
Photograph depicting extensive spontaneous abdominal wall hematoma and thigh hemorrhage in an older, previously unaffected man with an acquired factor VIII inhibitor.
-
Magnetic resonance image of an extensive spontaneous abdominal wall hematoma and thigh hemorrhage in an older, previously unaffected man with an acquired factor VIII inhibitor.
-
Coagulation Cascade
Tables
Classification |
Factor Activity, % |
Cause of Hemorrhage |
Mild |
>5-40 |
Major trauma or surgery |
Moderate |
1-5 |
Mild-to-moderate trauma |
Severe |
< 1 |
Spontaneous |
Indication or Site of Bleeding |
Factor level Desired, % |
FVIII Dose, IU/kg |
Comment |
Severe epistaxis; mouth, lip, tongue, or dental work |
20-50 |
10-25 |
Consider aminocaproic acid (Amicar), 1-2 d |
Joint (hip or groin) |
40 |
20 |
Repeat transfusion in 24-48 h |
Soft tissue or muscle |
20-40 |
10-20 |
No therapy if site small and not enlarging (transfuse if enlarging) |
Muscle (calf and forearm) |
30-40 |
15-20 |
None |
Muscle deep (thigh, hip, iliopsoas) |
40-60 |
20-30 |
Transfuse, repeat at 24 h, then as needed |
Neck or throat |
50-80 |
25-40 |
None |
Hematuria |
40 |
20 |
Transfuse to 40% then rest and hydration |
Laceration |
40 |
20 |
Transfuse until wound healed |
GI or retroperitoneal bleeding |
60-80 |
30-40 |
None |
Head trauma (no evidence of CNS bleeding) |
50 |
25 |
None |
Head trauma (probable or definite CNS bleeding, eg, headache, vomiting, neurologic signs) |
100 |
50 |
Maintain peak and trough factor levels at 100% and 50% for 14 d if CNS bleeding documented† |
Trauma with bleeding, surgery |
80-100 |
50 |
10-14 d |

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Approach Considerations
- Prehospital Care
- Emergency Department Care
- Factor VIII Concentrates
- Desmopressin
- Management of Bleeding Episodes by Site
- Treatment of Patients with Inhibitors
- Prophylactic Treatment
- Pain Management
- Complications
- Deterrence/Prevention
- Activity
- Gene Therapy
- Consultations
- Radiosynovectomy
- Show All
- Guidelines
- Medication
- Questions & Answers
- Media Gallery
- Tables
- References