Practice Essentials
Inherited deficiency of factor VII (FVII), the crucial enzyme triggering blood coagulation, is the most common of the rare coagulation disorders transmitted in an autosomal recessive manner. The clinical features are highly variable, ranging from severe (eg, intracranial or gastrointestinal hemorrhage) to milder (eg, epistaxis) to asymptomatic. [1]
On hemostatic testing, patients with factor VII deficiency have a normal aPTT and a prolonged PT; bleeding time is usually within the reference range. A specific assay for factor VII, using known factor VII–deficient plasma, is required to confirm the diagnosis. See Workup.
The choice of treatment for patients with factor VII deficiency depends on the site and severity of bleeding and the baseline factor VII activity. Therapeutic options for major bleeds include recombinant activated factor VIIa (rFVIIa), plasma-derived factor VII, fresh frozen plasma, and prothrombin complex concentrates. [2] Prophylaxis with rFVIIa may be used in patients who have experienced major bleeds. See Treatment and Medication.
Background
The discovery of vitamin K–dependent factors evolved slowly, after the initial identification of the role of prothrombin in blood clotting 100 years ago. In 1951, Alexander and colleagues identified factor VII as the key initiator of coagulation when they reported the first case of factor VII deficiency in a child and called it serum prothrombin conversion accelerator deficiency. [3]
Pathophysiology
Blood coagulation is a series of reactions in which plasma zymogens are converted into active enzymes. The final event of these reactions is the formation of an insoluble fibrin clot. These coagulant reactions are regulated by a number of stimulatory and inhibitory mechanisms. Thus, coagulation is a finely regulated system that maintains blood in a fluid phase but can rapidly respond to injury for the formation of clots. Factor VII is a vitamin K–dependent serine protease glycoprotein (also known as stable factor or proconvertin) with a pivotal role in hemostasis and coagulation. Other vitamin K–dependent factors include prothrombin, factors IX and X, and proteins C and S.
Tissue factor is an intrinsic membrane glycoprotein that is normally not exposed on the surface of intact blood vessels. When the vascular lumen is damaged, tissue factor is exposed and then binds to the small amounts of circulating factors VIIa and VII. This facilitates conversion of factor VII to factor VIIa. Factor VIIa bound to tissue factor in the presence of calcium and phospholipids facilitates the conversion of factor IX to factor IXa and factor X to factor Xa. Coagulation has traditionally been considered to occur via extrinsic and intrinsic pathways. Although this division is useful for understanding in vitro laboratory coagulation tests, no such division occurs in vivo because the tissue factor VIIa complex is a potent activator of factor IX and factor X.
Protein structure
Factor VII is synthesized in the liver and secreted as a single-chain glycoprotein of 48 kd. The epidermal growth factor domain has a calcium ion– binding site that to some degree mediates interaction with the tissue factor exposed at the site of vessel injury. Factor VII is then converted to factor VIIa. Gamma-glutamyl carboxylase catalyzes carboxylation of glutamine to Gla residues in the amino-terminal portion of the molecule. The carboxylase is dependent on a reduced form of vitamin K for its action. Whenever each glutamyl residue is carboxylated, the reduced vitamin K is converted to the epoxide form. Vitamin K epoxide reductase is required to convert the epoxide form of vitamin K back to the reduced form.
Warfarin inhibits the activity of vitamin K epoxide reductase and prevents recycling of vitamin K back to the reduced form, thus interfering with the synthesis of factor VII and other vitamin K–dependent factors. Warfarin poisoning can be reversed by administering vitamin K. Mutations of carboxylase can lead to low levels of all the gamma-carboxyglutamic acid domain-containing factors (ie, prothrombin; factors VII, IX, and X; protein C). [4]
Properties of factor VII
Factor VII is coded by the gene on band 13q34, closely located to the gene for factor X (F10). The plasma concentration of factor VII is 0.5 mg/L, and the plasma levels are determined by genetic and environmental factors. [5, 6] Factor VII has the shortest half-life of all procoagulant factors (3-6 h). Hence, when a problem with synthesis occurs, as in liver failure, vitamin K deficiency, or warfarin therapy, the factor VII level first decreases in the plasma, followed by a decrease in other vitamin K–dependent factors.
Factor VII levels are elevated during pregnancy in healthy females. Plasma factor VII levels also increase with age and are higher in females and in persons with hypertriglyceridemia. A strong contribution of the factor VII genotype to factor VII levels has been demonstrated, and different factor VII genotypes can result in up to several-fold differences in mean factor VII levels.
Activation
The major proportion of factor VII circulates in plasma in zymogen form, and activation of this form results in cleavage of the peptide bond between arginine 152 and isoleucine 153.
Rapid activation also occurs when factor VII is combined with its cofactor, which is the tissue factor in the presence of calcium (autocatalysis). This reaction may be initiated by a small amount of preexisting factor VIIa. Conversion of factor VII to factor VIIa is catalyzed by a number of proteases, including thrombin, factor IXa, factor Xa, factor XIa, and factor XIIa. Comparison of these proteins has shown that factor Xa, in association with phospholipids, has the highest potential to activate factor VII. [4, 7, 8]
Factor IXa is responsible for basal levels of plasma factor VIIa in healthy individuals. Patients with hemophilia B (factor IX deficiency), unlike patients with hemophilia A (factor VIII deficiency), have very low concentrations of circulating factor VIIa and achieve normal levels of VIIa within a few hours of infusion of purified factor IX.
Factor VIIa can be detected in plasma by a sensitive assay using a recombinant soluble form of tissue factor. The mean plasma concentration is 3.6 ng/mL in healthy individuals. The half-life of factor VIIa is relatively long (2.5 h) compared with other activated coagulation factors.
A summary of the structure and properties of coagulation factor VII is as follows:
-
Synthesis and localization - Synthesized in the liver and circulates in the plasma as a zymogen
-
Half-life - 3-6 hours
-
Molecular weight - 50,000
-
Structure - Amino-terminal (light chain) Gla domain, carboxy-terminal (heavy chain) catalytic domain, 2 epidermal growth factor domains
-
Cofactor - Tissue factor
-
Substrate - Factor VIIa/tissue factor complex activates factors X and IX
Role of factor VII in coagulation and coagulation pathways
The association of factor VIIa with tissue factor enhances the proteolytic activity by (1) bringing the binding sites for both the substrate (factors X and IX) and the enzyme (VIIa) into closer proximity and by (2) inducing a conformational change, enhancing the enzymatic activity of factor VIIa.
The factor VIIa/tissue factor complex formed as a result of binding of small amounts of preexistent plasma factor VIIa activates factor X and factor IX. The rate of factor X activation by this pathway (extrinsic) is approximately 50 times slower than the rate achieved by factor IXa, factor VIIIa, phospholipid, and calcium ions (intrinsic pathway). Factor Xa formed by both enzyme complexes binds to membrane-bound factor Va to produce the prothrombinase complex. This complex converts prothrombin to thrombin, which results in the formation of fibrin clots. Note the image below.
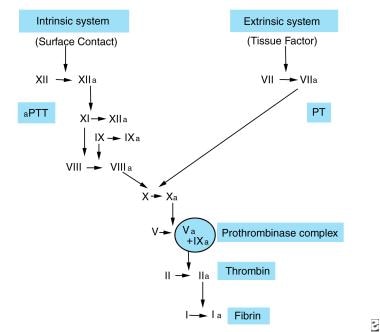 Factor VII. Intrinsic and extrinsic pathways of coagulation. Factor VII/tissue factor complex activates factor IX and factor X. Factor IXa along with factor VIIIa results in formation of more factor Xa. Factor Xa along with factor Va converts prothrombin to thrombin.
Factor VII. Intrinsic and extrinsic pathways of coagulation. Factor VII/tissue factor complex activates factor IX and factor X. Factor IXa along with factor VIIIa results in formation of more factor Xa. Factor Xa along with factor Va converts prothrombin to thrombin.
Inhibition of the extrinsic pathway of coagulation
Activation of factor X by the factor VIIa–tissue factor complex results in the interaction of factor Xa with factor Va to form a prothrombinase complex. Very small amounts of thrombin formed during this initiation phase of thrombin generation subsequently activate platelets, factor VIII, factor V, and factor XI. This leads to the propagation phase, wherein the bulk of the thrombin is generated. The initiation and propagation phases of the coagulation system are differentially regulated by the inhibitors. Tissue factor pathway inhibitor targets factor VIIa/tissue factor/factor Xa product complex and principally serves to regulate the initiation phase of the reaction.
The antithrombin III/heparin complex plays a major role in the inhibition of all vitamin K–dependent proteases except factor VIIa.
Factor VII deficiency
Because factor VII deficiency is a rare disease, data concerning the pathophysiology are limited. Both qualitative and quantitative forms of factor VII deficiency have been noted. Factor VII Padua I has been described in one kindred with an abnormal rabbit brain prothrombin time (PT) but a normal ox brain PT; factor VII (Verona) is associated with an abnormal form of factor VII, and kindreds with heterozygosity for this type have been reported. Factor VII Padua 2 is a double-heterozygote condition associated with abnormal coagulation test results with only ox brain thromboplastin.
Over 220 different mutations have been identified since the isolation of the factor VII gene (F7). Most described mutations are missense mutations. Nonsense mutations, small deletions, and splice-site abnormalities have also been identified. A few large genomic rearrangements and six common variants have been identified. Nevertheless, in spite of an exhaustive direct sequencing of F7 exons and exon-intron junctions and of the proximal promoter region, a significant proportion of defective alleles has not been identified yet. The rate of uncharacterized F7 disease alleles ranges from 2% to 8% in Europe, and a similar estimate (7%) was made in India. [1]
Factor VII coagulant activities measured in the laboratory are not well correlated with bleeding manifestations. [9] This is partly because different F7 mutations express different levels of coagulant activity. Additionally, factor VII activity levels are variable when assayed in the presence of tissue factor obtained from different species.
Approximately two thirds of the mutations seem to affect the protease domain, indicating that loss of protease function is the most common cause of the clinical phenotype. [9]
The donor splice mutation in intron 7 (IVS7+7) was first described in Italy. Ala294Val and Ala294Val;404delC was first described by Arbini et al in Polish patients and by Bernardi et al in Italian patients. [10] According to Herrmann et al, this was found to be the most common type of mutation in Europe. [9] In the same study, homozygous conditions to mutations Val (-17) Ile, Phe4Leu, Cys135Arg, Ala244Val, Ala294Val;404delC, and IVS4+1G>A were associated with factor VII activities of 8%, less than 1%, 1-4%, 3%, less than 1%, and 7%, respectively. Factor VII activities ranging from 75-80% were found in heterozygous patients with donor splice mutation IVS7+7, which is thus considered a mild mutation. [9]
Factor VII activity is influenced by mutations of F7 and by allelic polymorphic variations of the gene. Eight polymorphisms within F7 are known, 3 of which (ie, an insertion polymorphism of the promoter, a repeat polymorphism within intron 7, the Arg353Gln polymorphism of exon 8) influence the level of factor VII activities. Analysis of 7 of the polymorphisms in 14 patients showed only a mild decrease (> 50%) of factor VII levels in those without an identified mutation compared with those with an identified mutation. These data appear to indicate that patients with activated factor VII levels greater than 50% are less likely to have a definitive F7 mutation, although polymorphisms of the F7 gene can be detected in these patients. [11]
A detailed database of mutations is available at the MRC Haemostasis & Thrombosis Database Resource Site.
Epidemiology
Frequency
International
Hereditary factor VII deficiency is a rare autosomal recessive bleeding disorder first described by Alexander et al in 1951. [3] Prevalence is estimated to be 1 case per 500,000 persons in the general population. Dubin-Johnson syndrome and Rotor syndrome are associated with a high prevalence of factor VII deficiency. [12]
Acquired factor VII deficiency from inhibitors is very rare. Cases have been reported in association with drugs such as cephalosporins, penicillins, and oral anticoagulants. Acquired factor VII deficiency has also been reported to occur spontaneously or with other conditions, such as myeloma, sepsis, and aplastic anemia, and with interleukin-2 therapy and antithymocyte globulin therapy.
Race
Specific mutations and polymorphisms are known to occur in some populations. Among Iranian and Moroccan Jews, a missense Ala244Val mutation in the F7 gene is responsible for many cases of factor VII deficiency. A study of residents of southern Israel, consisting primarily of Jews of North African descent, Ashkenazi Jews, and Bedouins, found an incidence of 1:13,000 for factor VII deficiency (activity under 60%) and 1:40,000 for severe factor VII deficiency (activity under 10%). The majority of severe cases were found among Bedouin residents with a high prevalence of consanguineous marriage. [13]
The highest frequencies of Arg353Gln substitution, an F7 polymorphism that results in decreased factor VII levels, are observed in Gujaratis (25%) and Dravidian Indians (29%), compared with northern Europeans (9%) and Japanese (3%), . [14]
Prognosis
Morbidity and mortality rates vary with the severity of the factor deficiency. Factor VII deficiencies < 1% result in bleeding disorders indistinguishable from severe hemophilia A or hemophilia B. Recombinant agent therapy and early intervention of joint disease may result in improved outcomes, as in persons with hemophilia A.
Patient Education
Patients and family members should be educated about the disease and its transmission. Genetic counseling is recommended. Advise patients to seek medical attention early during the development of symptoms, to consult with specialists, and to comply with follow-up requirements.
For patient education resources, see Hemophilia.
-
Factor VII. Intrinsic and extrinsic pathways of coagulation. Factor VII/tissue factor complex activates factor IX and factor X. Factor IXa along with factor VIIIa results in formation of more factor Xa. Factor Xa along with factor Va converts prothrombin to thrombin.