Practice Essentials
Immunoglobulin-related amyloidosis is a monoclonal plasma cell disorder in which the secreted monoclonal immunoglobulin protein forms insoluble fibrillar deposits in one or more organs. In nearly all cases, the deposits contain immunoglobulin light (L) chains or L-chain fragments, termed L chain–type amyloidosis (AL).
In a few reported patients, the amyloid deposits have contained immunoglobulin heavy (H) chains; these are termed H chain–type amyloidosis AH). Before the discovery that the major fibril component in these patients was an immunoglobulin fragment, patients with light chain–type amyloidosis were described as having primary amyloidosis (primary in the sense of idiopathic) or, when the burden of monoclonal plasma cells was large, myeloma-associated amyloidosis.
Immunoglobulin L and H chains are two of 20 different fibril proteins that have been described in human amyloidosis. For a general discussion of the human amyloidoses, the types of human amyloidosis, and an approach to the diagnosis of amyloidosis, see Amyloidosis.
L chain–type (AL) amyloidosis is related to both multiple myeloma and monoclonal gammopathy of undetermined significance (MGUS). These monoclonal plasma cell disorders can be categorized according to the total body burden of monoclonal plasma cells. When this burden is large, the diagnostic criteria for multiple myeloma are fulfilled; when this burden is lower, MGUS is diagnosed. Multiple myeloma and MGUS fall on a continuum, with 20% of patients with MGUS progressing to multiple myeloma within 10 years.
In most patients with a monoclonal plasma cell disorder, whether multiple myeloma or MGUS, the monoclonal L chain secreted by the clone remains soluble in the bloodstream. However, in some patients, the physicochemical characteristics of the immunoglobulin L chain or L-chain fragment lead to its deposition as amyloid. Thus, some patients with AL amyloidosis meet the diagnostic criteria of multiple myeloma, whereas other patients can be considered as having MGUS in which the clonal immunoglobulin product is amyloidogenic.
In addition to cases of monoclonal gammopathy in which the secreted clonal immunoglobulin remains in solution and those in which secreted clonal immunoglobulin forms amyloid deposits, a third group consists of cases in which the monoclonal proteins accumulate in various organs, but the deposits do not form fibrils. Patients with this form are described as having nonamyloid monoclonal immunoglobulin deposition disease (MIDD). The relationship among the plasma cell dyscrasias and the amyloidoses is depicted in the image below.
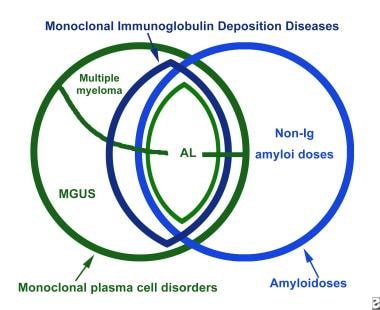 The relationship among light chain–type amyloidosis (AL), the other monoclonal plasma cell disorders, and the other amyloidoses. Ig = immunoglobulin; MGUS = monoclonal gammopathy of undetermined significance.
The relationship among light chain–type amyloidosis (AL), the other monoclonal plasma cell disorders, and the other amyloidoses. Ig = immunoglobulin; MGUS = monoclonal gammopathy of undetermined significance.
Definitive diagnosis of all forms of amyloidosis is by a positive Congo red stain of a biopsy specimen. Currently, specimens are obtained by aspiration of subcutaneous abdominal fat. (See Workup.) Current choices for first-line treatment of systemic L-chain amyloidosis are autologous stem cell transplantation (ASCT) and combination chemotherapy (see Treatment and Medication).
Pathophysiology
The most common light chain–type amyloidosis precursor proteins are L chains of the lambda (l) class. The lambda light chain–type amyloidosis is approximately twice as prevalent as the kappa (k) light chain–type amyloidosis, and L chains of the Vl 6 class are the most amyloidogenic. Clonal plasma cell proliferative diseases in which the Vl 6 gene is expressed are always associated with amyloid deposition. Among Vk genes, the Vk 1 subgroup is overrepresented among amyloid-forming L chains.
Within the V region families, certain amino acid residues occurring at particular positions in the L-chain sequence render those chains more amyloidogenic, with a combination of such residues increasing the chances of a particular L-chain protein being associated with tissue amyloid deposition. Another structural feature that appears to predispose to L chain – type amyloid deposition is enzymatic glycosylation of the L chain. Although 15% of human L chains bear sugar residues, almost one third of amyloidogenic L chains are glycosylated. Why certain amino acid and glycosylation characteristics in L chains predispose to amyloid formation remains unknown.
L-chain–type amyloid deposits contain intact L chains; L-chain fragments; or, in most patients, both. The fragments always include the amino terminus of the chain and range in mass from 5000 to 16,000 d. In 90% of patients, the deposited peptides include at least some constant region sequence; therefore, the peptides react with commercially available anti–L chain sera, which are specific for constant region determinants. These observations explain why 10% of deposits do not bind either commercial anti-k or anti-l antisera.
L-chain–type amyloid deposits can develop in any organ system. The most common organs involved are the kidneys, the heart, the gastrointestinal (GI) tract, the peripheral nerves, and the liver. In most cases, the deposits affect multiple organ systems. Factors leading to the specific pattern of organ involvement in a particular patient are not understood.
In a minority of cases, localized amyloid deposits, including amyloid masses (amyloidomas), may be found in various sites, even in the absence of systemic disease. The pathogenesis of localized light chain–type amyloidosis is not well understood, but a small, localized clone of plasma cells apparently produces immunoglobulin, which forms deposits near the site of synthesis. In some patients, plasma cells have been demonstrated histologically, accompanying the localized amyloid deposits. In one patient, DNA sequencing revealed that local plasma cells were producing the locally deposited L chains. [1]
Etiology
No cause is known for any of the monoclonal plasma cell dyscrasias. Some evidence supports an etiologic role for human herpesvirus 8 (HHV-8), but this proposed etiology remains controversial. Other suggested risk factors include genetic predisposition, environmental or occupational exposures, and chronic inflammation.
Epidemiology
United States
Annually, one to five cases of immunoglobulin-related amyloidosis per 100,000 people occur. The best available direct data on the frequency of L chain–type amyloidosis in the United States come from Olmsted County, Minnesota, where the incidence rate of L chain–type amyloidosis was calculated to be 1.2 per 100,000 person-years. [2] The population in this location is primarily of northern European ancestry. Whether this finding applies to different populations is not known.
Based on indirect calculations, the frequency may be higher. The annual incidence of multiple myeloma is approximately five cases per 100,000 people, and the prevalence of L chain–type amyloidosis in patients with myeloma is approximately 20-35%, producing an overall incidence of combined L chain–type amyloidosis and myeloma of 1-1.5 cases per 100,000 people. Only one in five patients with L chain–type amyloidosis has frank myeloma; therefore, the total number of patients with L chain–type amyloidosis type is five times the number of patients with L chain–type amyloidosis and myeloma, or at least five cases per 100,000 people.
International
The international incidence of amyloidosis is not well documented, but has been estimated at 5 to 13 cases per million population per year. L chain–type amyloidosis is the most prevalent type, accounting for 65% of cases in the United Kingdom and 93% of cases in China. [3]
Race-, Sex-, and Age-related Demographics
L chain–type amyloidosis affects people of all racial and ethnic groups. No data are available comparing the incidence of disease in different groups.
The male-to-female incidence ratio of L chain–type amyloidosis is 2:1.
The median age at diagnosis of immunoglobulin-related amyloidosis in the largest published series (from the Mayo Clinic) was 64 years. [4]
Prognosis
The prognosis for patients with L chain–type amyloidosis depends largely on the specificity of the tissue deposition. Any organ can be involved, with symptoms and physical findings reflecting the pattern of anatomic compromise. Factors that cause deposits in different organs in different patients are unknown. Cardiac deposition is the most severe consequence of systemic L chain–type amyloidosis, eventually occurring in most patients, and is the most common cause of death in those patients.
Patients with clinical cardiac involvement have the worst prognosis, with a median survival rate of 6 months. In these patients, increased troponin is associated with worse left ventricular and left atrial functions. Increased troponin at baseline strongly predicts all-cause mortality. [5]
Median overall survival was 29 months in a retrospective study of 63 patients with L chain–type amyloidosis, of whom 32 (51%) presented with cardiac amyloid involvement. Overall survival did not differ between patients with or without cardiac involvement. During a median follow-up of 12.7 months, 38 (60%) patients died. [6]
Patients with involvement limited to the peripheral nerves have the longest survival. Other favorable prognostic features include a small number of clonal plasma cells in the bone marrow and normal kidney function.
In the absence of chemotherapy, systemic L chain–type amyloidosis is always progressive. A subgroup of cases respond to chemotherapy with temporary resorption of amyloid fibrils and improvement of end-organ function.
In a retrospective study of 146 patients with L chain–type amyloidosis that relapsed after treatment with chemotherapy and autologous stem cell transplant, median overall survival and 5-year overall survival from the time of post-transplant relapse were 51.7 months and 39%, respectively. [7]
In a single-institution review of 1551 patients with newly diagnosed AL amyloidosis seen from 2000 to 2014, Muchtar et al reported that overall, outcomes in these patients improved over that period, with earlier diagnosis, higher rates of very good partial response, lower early mortality, and improved overall survival. Four-year overall survival in the autologous stem cell transplant (ASCT) population showed the greatest gains after 2010, rising to 91% from 65%. In the non-ASCT group the greatest gains were after 2005, rising to 38% from 16%. [8]
-
The relationship among light chain–type amyloidosis (AL), the other monoclonal plasma cell disorders, and the other amyloidoses. Ig = immunoglobulin; MGUS = monoclonal gammopathy of undetermined significance.






