Practice Essentials
Regardless of the etiology, a deficiency of cyclooxygenase (COX), a key regulatory enzyme in the synthetic pathway of eicosanoid production, results in beneficial and detrimental physiologic conditions relative to imbalances of the eicosanoids. [1] Thus, tracing research of the COX pathway is essential to an understanding of COX deficiency, and examining the variable effects of COX inhibition are advantageous.
Eicosanoids, which include prostaglandins, leukotrienes, thromboxanes, and lipoxins, are responsible for multiple inflammatory, mitogenic, and angiogenic activities in various tissue and organ systems. Therefore, COX — also known as prostaglandin-endoperoxide synthase (PTGS), fatty acid COX, prostaglandin H (PGH) synthase, and EC 1.14.99.1 — is implicated in the production of fever, inflammation, and pain.
In 1930, American gynecologists Kurzok and Lieb first described the stimulatory effects of seminal fluid on human uterine muscle tissue. A few years later, von Euler of Sweden independently discovered similar effects of human seminal fluid on smooth muscle tissue. He localized the biologic activity to a fraction of lipid soluble acids that he termed "prostaglandin," hypothesizing that these substances originate in the prostate gland. Two decades later, the prostaglandins were deduced to be a family of related compounds that contain 20-carbon polyunsaturated fatty acids with a cyclopentane ring, as depicted below.
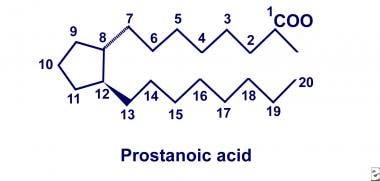
By 1964, after recognition of this basic structure, Bergstrom and colleagues successfully synthesized series 2 prostaglandins from arachidonic acid using sheep seminal fluid. However, the physiologic significance of prostaglandin production did not unfold until 1971, when Vane, Smith, and Willis discovered that aspirin and indomethacin inhibited prostaglandin biosynthesis. Further investigations by Smith concluded that aspirin and indomethacin inhibited synthesis by specifically blocking oxygenation of arachidonic acid. Collectively, these landmark discoveries provided initial insight into the COX pathway of arachidonate metabolism.
Advances in genomic analysis have led to a clearer understanding of the COX pathway. Initial investigations by Miyamoto and Simmons demonstrated that 2 isoforms exist: COX-1 (PTGS-1) and COX-2 (PTGS-2), respectively. The transcription of COX-1 yields a 2.7-kilobase (kb) messenger ribonucleic acid (mRNA) that encodes a 576-residue, 65-kd protein. Conversely, the transcription of COX-2 yields a 4.5-kb mRNA that encodes a 70-kd protein with roughly 70-75% homology to the COX-1 protein.
Funk and co-investigators localized COX-1 to 9q32-q33.3 via somatic hybrid deoxyribonucleic acid (DNA) analysis. Tay and colleagues then localized COX-2 to 1q25.2-q25.3 via fluorescein in situ hybridization. Furthermore, using sequence analysis of human genomic DNA, researchers concluded that the amino acids important for catalysis by COX-1 are conserved and are equally important for catalysis by COX-2.
Activity of COX-1 and COX-2
Evidence suggests that COX-1 and COX-2 are similar in structure and function but that they exist as 2 distinct enzymatic entities. They have been defined as monotropic integral membrane proteins located primarily in the endoplasmic reticulum (COX-1) and the perinuclear envelope (COX-2). Their distinct biosynthetic activity includes an endoperoxidase synthase reaction that oxygenates and cyclizes polyunsaturated fatty acid precursors (eg, arachidonic acid) to form prostaglandin G2 (PGG2), and a peroxidase reaction that converts PGG2 to prostaglandin H2 (PGH2), as shown below. In turn, PGH2 is converted to biologically active products (ie, prostaglandin E2 [PGE2]) by individual synthase and reductase reactions.
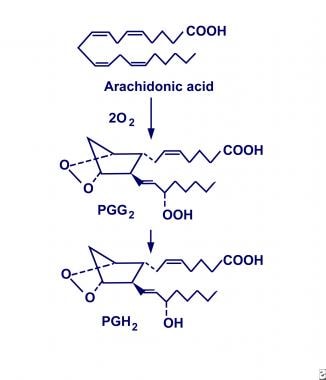
COX-1 is expressed constitutively and is isolated throughout most cell lines in almost all mammalian tissues. It is described as a housekeeping enzyme, being responsible for cell-to-cell signaling, tissue homeostasis, and cytoprotection. In view of this, researchers hypothesize that COX-1 is a rapid responder to various physiologic conditions.
Conversely, COX-2 is described as an inducible isoform influenced by a plethora of proinflammatory mediators. Without appropriate stimulation, isolation of the COX-2 protein is negligible in most tissues. However, newer literature reveals that COX-2 is expressed constitutively in some cell lines of the brain, kidney, and trachea. Although vaguely described, COX-2 is considered to be a principal mediator of inflammation, mitogenesis, and angiogenesis.
Studies have demonstrated that the eicosanoids produced by cytosolic COX-1 participate in autocrine and paracrine activities, while those produced by perinuclear COX-2 result in intracrine activity. This subcellular localization of the COX enzyme (through the use of new microscopy techniques) has helped to explain why 2 isoforms exist. Furthermore, researchers have proposed that COX-1 and COX-2 acquire arachidonic acid from different phospholipases, suggesting participation through separate pathways.
Etiology
COX deficiency typically denotes an acquired cause-and-effect relationship between the protein's enzymatic activity and nonsteroidal anti-inflammatory drugs (NSAIDs). However, Nyman and co-investigators, while studying bleeder families on the Aland Islands, reported an autosomal dominant pattern of inheritance for COX deficiency. [2, 3]
In addition, Horellou and colleagues described 3 family members of 2 successive generations with bleeding tendencies and concluded that their platelets were compatible with a COX deficiency. [4] This study also described an autosomal dominant pattern of inheritance but deduced that COX was not imperative to aggregation and adenosine triphosphate (ATP) release in the face of high collagen concentrations. Sporadically documented familial and constitutional bleeding disorders have been associated with platelet COX-1 deficiency.
Pathophysiology
Two types of platelet COX-1 deficiency have been recognized. The type 1 defect is characterized by a complete absence of COX-1 protein in platelets. In the type 2 deficiency, on the other hand, a normal level of protein exists, but COX-1 enzymatic activity is impaired. The physiologic consequences of COX deficiency, however, are best described as acquired, rather than inherited, disorders.
The effect of COX-1 deficiency varies from tissue to tissue and largely is determined by the fate of the end products. Several studies have concluded that direct inhibition of COX-1 coincides with a loss of cytoprotection. In turn, COX-1 deficiency has been deemed as a significant participant in gastric and renal pathology. [5]
However, Langenbach and colleagues reported that mice deficient in COX-1 live uneventful lives despite a 99% reduction in overall prostaglandin production. [6] He also emphasized that a lack of gastric and renal pathology existed in spite of observed prostaglandin deficiency. Confirming earlier literature, Langenbach reported that female mice deficient in COX-1 had difficulties with parturition, while male mice deficient in COX-1 were unimpaired. Langenbach also has demonstrated that, contrary to popular belief, COX-1 deficiency correlates with reduced edema in the face of inflammatory mediators that increase arachidonate, illuminating its significance in the inflammatory cascade.
Without significant controversy, the consensus has been that the inhibition of COX-2 is responsible, in part, for the antipyretic, analgesic, and anti-inflammatory properties of NSAIDs. However, novel investigations using genetically altered COX knockout mice have uncovered new ramifications associated with COX-2. First, Lim and colleagues observed that COX-2–deficient female mice incurred reproductive failures via abnormalities of ovulation, fertilization, implantation, and decidualization, although follicular development was normal. [7]
Independently, Dinchuck and coauthors described reduced neonatal viability in mice deficient in COX-2, as a consequence of renal dysplasia, cardiac fibrosis, infertility, and endotoxin-induced, hepatocellular cytotoxicity. [8] He concluded that COX-2 deficiency does not affect viability in utero and that heterozygous and wild-type mice do not have disease. Cheng and colleagues subsequently cited the importance of COX-2 as a "mediator of increased renin production in response to inhibition of angiotensin II production." [9] As a result, they proposed that a component of the renin-angiotensin feedback system may be modulated by COX-2 expression.
Through the evolving study of COX, new theories have been integrated into previously well-described pathways. Nevertheless, the influence that COX has on each cell, tissue, organ, and system results from precursor, product, and receptor variability.
Precursors
The eicosanoids, which include prostaglandins, leukotrienes, thromboxanes, and lipoxins, are derived from the oxygenation of 20-carbon polyunsaturated essential fatty acids via the COX and lipoxygenase pathways. [1, 10] However, only a fraction of these 20-carbon polyenoic acid precursors are the substrates that actually yield eicosanoids. The most significant of the precursors include the following:
-
8,11,14-eicosatetraenoic acid (dihomo-g-linolenic acid)
-
5,8,11,14-eicosatetraenoic acid (arachidonic acid)
-
5,8,1,14,17-eicosapentaenoic acid
Omega-6 fatty acids, the essential substrates of dihomo-g-linolenic acid and arachidonic acid, are derivatives of linoleic acid. Omega-3 fatty acids, the essential substrates of eicosapentaenoic acid, are derivatives of a-linolenic acid. As a result, linoleic and linolenic fatty acid pathways of desaturation and elongation provide the essential precursors for COX and lipoxygenase that ultimately result in the production of eicosanoids. [10]
Products
As previously noted, COX metabolizes arachidonic acid (5,8,11,14-eicosatetraenoic acid) into PGH2 intermediates. Subsequently, PGH2 is converted into biologically active products via cell-specific enzymatic reactions. The products include not only classic prostaglandins (ie, prostaglandin D2 [PGD2], PGE2, prostaglandin F2-alpha [PGF2-alpha]), but also prostacyclin (PGI2) and thromboxane A2 (TXA2). [11]
Besides these dienoic products, COX metabolizes analogous fatty acids into monoenoic and trienoic prostaglandins and thromboxanes as a consequence of the number of precursor double bonds. Collectively, their diverse properties are implicated in several physiologic processes as receptor-dependent mediators and as intracellular secondary messengers. However, the potencies of different monoenoic, dienoic, and trienoic prostaglandins and thromboxanes vary with respect to each family. Studies emphasize that the products are particularly influential in local biologic environments, because of their rapid conversion to inactive metabolites. Hence, precursor availability and enzyme kinetics play a key role in the regulation of individual responses.
Thromboxane A
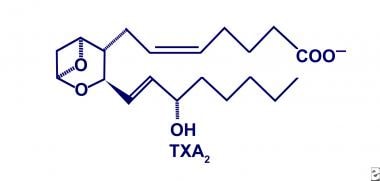
TXA2, the predominate product of COX in platelets and macrophages, is converted from PGH2 by thromboxane synthase. The structural characteristics include a 6-membered ring containing an ether, as depicted in the image below. The functional characteristics include platelet aggregation, vascular smooth muscle constriction, and bronchial smooth muscle constriction with a corresponding 30-second tissue half-life. Furthermore, TXA2 has a different affinity for each eicosanoid-specific receptor because of distinct receptor ligands. As a result, the physiologic responses via TXA2 are tailored to the situation and are less haphazard when stimulated. [12]
Prostacyclin
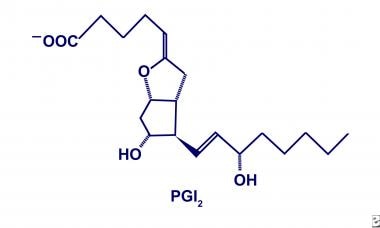
PGI2, the predominate product of COX in microvascular endothelium, is converted from PGH2 by PGI2 synthase. The structural characteristics include not only a 5-member ring, similar to all prostaglandins, but also an enzyme-specific arrangement of respective hydroxyl and carbonyl groups, as shown below. The functional characteristics include inhibition of platelet aggregation, inhibition of platelet and neutrophil adhesion, dilation of bronchial and vascular smooth muscle, and modulation of cholesterol efflux from arterial walls with a corresponding 3-minute tissue half-life. Furthermore, the biosynthesis of PGI2 is enhanced in the face of thrombogenesis and vasoconstriction to balance the physiologic milieu, not unlike several well-known stimulation/inhibition processes. [11]
Prostaglandin D
PGD2, the predominate product of COX in mast cells, is converted from PGH2 by endoperoxide-D isomerase. Again, the structural characteristics are similar to those of all other prostaglandins. The functional characteristics include symptoms associated with histamine release (eg, hypotension) and poorly defined roles associated with immunologic processes. However, the formation of 9-alpha,11-beta-prostaglandin F (PGF) metabolites by preferential conversion of PGD2 results in the inhibition of platelet aggregation and the contraction of vascular smooth muscle tissue. This might explain the occasional hypertensive patient observed with systemic mastocytosis.
Prostaglandin E
PGE2 is a significant product of COX in the gastric mucosa, renal medulla, and microvascular endothelium, as well as in some tumors. [13, 14] This biologically active product is converted from PGH2 by endoperoxide-E isomerase. Although the products are structurally related, each functional temperament is curiously diverse. (See the image below.)
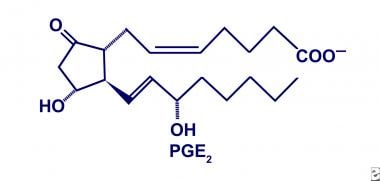
In the gastric mucosa, PGE2 preserves integrity by influencing mucus and bicarbonate secretion. [15] It also maintains mucosal blood flow and participates in cellular repair. In the renal medulla, PGE2 enhances vasodilatation and inhibits tubular sodium absorption. [5] Hence, a deficiency of PGE2, as observed in essential hypertension, results in unopposed vasoconstriction.
Further attributes include stimulation of local osteoclasts, relaxation of bronchial smooth muscle tissue, contraction of uterine smooth muscle tissue, and modulation of presynaptic adrenergic neuron receptors. Despite vague interpretation, PGE2 is hypothesized to also be a key participant in local inflammatory responses. [16, 17]
COX Expression and Platelet Function
Platelets have 3 distinct roles in coagulation: (1) initial adhesion, (2) phospholipid externalization, and (3) platelet aggregation.
Research suggests that TXA2, the final product of platelet arachidonic acid metabolism, is necessary for normal platelet function. Aggregation occurs as TXA2 diffuses from the platelet and binds to glycoprotein IIb-IIIa (GPIIb-IIIa) platelet membrane receptors to enhance alpha-granule and dense-granule secretion. The alpha granules store platelet activation markers, whereas the dense granules store calcium, serotonin, and ATP. Thus, inhibition of COX diminishes platelet secretion and inevitably disrupts normal platelet function.
Platelets are unique in that they do not regenerate COX. This becomes apparent in the face of irreversible inhibitors, including, notoriously, aspirin. [18] COX inhibition by aspirin results in diminished TXA2 production and, inevitably, in the loss of platelet aggregatory properties for the life of the platelet (ie, 7-10 days). [19, 20]
However, studies have demonstrated that high concentrations of strong promoters (eg, thrombin, collagen) are not dependent on TXA2. In fact, it has been reported that total inhibition of platelet COX does not alter a complete platelet response, suggesting that multiple pathways of platelet activation exist. [21] Perhaps this observation accounts for the normal hemostasis observed in persons who are taking aspirin.
Inherited disorders of platelet function are diverse and probably underreported. One of these disorders is the aspirinlike defect (ALD), which produces a mild impairment in hemostasis. ALD is a rare, inherited autosomal dominant dysfunction of the intraplatelet arachidonic acid pathway, leading to impaired TXA2 signalling. ALD should be suspected when platelet-type bleeding symptoms are present. Platelet aggregation studies can be performed to identify ALD; if ALD is present, family studies are important for identifying individuals who may be at risk of increased hemorrhage during surgical procedures. [22]
COX Expression and Colon Carcinoma
COX expression has been implicated in colon carcinoma since epidemiologic studies first described reduced mortality rates in subjects taking aspirin. [23, 24] Initially, Tsujii and DuBois studied the effects of COX-2 overexpression by transfecting rat intestinal epithelium. [25] They demonstrated that overexpression increased adhesion and inhibited apoptosis, perhaps enhancing tumorigenic potential. [13]
In a study in which Oshima and colleagues bred mice with APC mutations similar to those of humans, COX was uncovered as a significant factor in colon carcinoma. [26, 27] The investigators reported that mice carrying an APC mutation and who were wild type for COX-2 expression developed an average of 652 polyps at 10 weeks, whereas heterozygous mice developed 224 polyps, and null mice developed 93 polyps. They also found that drugs that inhibited COX-2 (but not COX-1) greatly reduced polyp formation.
Further research by Langenbach and coauthors, using multiple intestinal neoplasia mice (which led to 100% incidence of carcinoma), showed that COX-1 and COX-2 deficiency decreased polyps by 70-80%. [6] Although a correlation exists between COX expression and colon carcinoma, the mechanism of action remains unknown. [28] Moreover, the described studies may demonstrate only one of many antitumorigenic pathways that, irrespective of COX dependence, are affected by NSAIDs. [29, 30, 31]
Nevertheless, studies have shown that NSAIDs, including selective COX-2 inhibitors (celecoxib), augment the sensitivity of many kinds of tumors toward chemotherapeutics. [32] In that regard, celecoxib and 5-fluorouracil used in combination have a synergistic antitumor effect in a colon cancer mouse model. The mechanism of this interaction may be activation of an apoptosis signal. [33]
COX Inhibition by NSAIDs
Once Vane and colleagues deduced that aspirin and indomethacin inhibited the COX pathway of arachidonate metabolism, numerous investigations were performed to better understand the mechanism by which NSAIDs interacted with COX. [34, 35, 36] Smith and Dewitt explained fundamental differences among the NSAIDs based on their interaction with COX active sites. [37] Consequently, they described 3 distinct classes of inhibitors: class I, II, and III NSAIDs. [38]
Class I NSAIDs
Defined as simple, competitive COX inhibitors, Class I NSAIDs include ibuprofen, piroxicam, naproxen, mefenamic acid, sulindac sulfide, and flufenamic acid. They interact rapidly and reversibly with COX active sites to form an enzyme-inhibitor (EI) complex. Smith and Dewitt determined the inhibitory concentration 50% (IC50) of several class I NSAIDs and observed that an overall higher affinity for the COX-1 isoform exists. [39] However, they emphasized that the relative potency of individual class I NSAIDs may vary between in vitro and in vivo models.
Class II NSAIDs
Class II NSAIDs, which include indomethacin, flurbiprofen, meclofenamate, and diclofenac, are defined as competitive, time-dependent COX inhibitors. Their interaction with COX active sites is laggard relative to class I inhibitors. After initial formation of the EI complex, the inhibitor causes a slow, but reversible, conformational change in the protein to form an EI* complex. This non–covalently bound, semistable complex is less susceptible to proteases and dissociates from COX active sites at a slower rate than do EI complexes.
Several studies have indicated that different class II inhibitors possess different k2 and k-2 values for COX-1 and COX-2 active sites based on in vivo selectivity. Although the biochemical mechanism of EI* complex formation is unknown, researchers attribute the kinetic properties of class II inhibitors to structural differences.
Class III NSAIDs
Class III NSAIDs include aspirin, salicylic acid, and valeryl salicylate and are defined as irreversible, competitive, time-dependent COX inhibitors. Although these agents are categorized together, only aspirin covalently modifies COX-1 and COX-2. This modification results after aspirin binds to the COX active site and transfers its acetyl group to a definitive serine residue. The acetylation of COX-1 specifically interferes with the endoperoxidase reaction that normally oxygenates and cyclizes arachidonic acid to form PGG2. Acetylation of COX-2 results in isoform modification that yields 15R-hydroxyeicosatetraenoic acid (15R-HETE) instead of PGG2. [40]
Questions & Answers
Overview
What is cyclooxygenase (COX) deficiency?
What is the physiology of cyclooxygenase 1 (COX-1) and cyclooxygenase 2 (COX-2)?
What causes cyclooxygenase (COX) deficiency?
What is the pathophysiology of cyclooxygenase (COX) deficiency?
What is the role of eicosanoids in the pathophysiology of cyclooxygenase (COX) deficiency?
What are the products produced by cyclooxygenase (COX)?
What is the role of cyclooxygenase (COX) deficiency in platelet dysfunction?
What is the role of cyclooxygenase (COX) deficiency in colon carcinoma?
What is the role of NSAIDs in the etiology of cyclooxygenase (COX) deficiency?
What is the role of class III NSAIDs in the etiology of cyclooxygenase (COX) deficiency?
What is the role of class I NSAIDs in the etiology of cyclooxygenase (COX) deficiency?
What is the role of class II NSAIDs in the etiology of cyclooxygenase (COX) deficiency?
-
Twenty-carbon polyunsaturated fatty acid with cyclopentane ring.
-
Cyclooxygenase conversion of arachidonic acid into prostaglandin H2 (PGH2).
-
Thromboxane A2 (TXA2).
-
Prostacyclin (PGI2).
-
Prostaglandin E2 (PGE2).






