Practice Essentials
Glomus jugulare tumors are rare, slow-growing, hypervascular tumors that arise within the jugular foramen of the temporal bone and frequently involve the lower cranial nerves. [1] They are included in a group of tumors referred to as paragangliomas, which occur at various sites and include carotid body, glomus vagale, and glomus tympanicum tumors. [2] Jugular foramen syndrome, or paresis of cranial nerves IX to XI, is pathognomonic for these tumors. [3]
Paragangliomas are tumors originating from the paraganglionic system (autonomic nervous system), mostly found at the region around the jugular bulb; thus, they are also referred to as glomus jugulare tumors. Although these lesions appear to be histologically benign, they clinically present with great morbidity, especially due to invasion of nearby structures such as lower cranial nerves. These tumors are challenging to manage, as treatment involves complex approaches and requires great knowledge of the skull base. [4]
Glomus tumors are rare, vascular, slow-growing tumors, and most are benign [3] Glomus tumors are also referred to as chemodectomas or nonchromaffin paragangliomas. Paragangliomas are often found at other sites, including the middle ear (glomus tympanicum tumor), the carotid body (carotid body tumor), and the vagus nerve in proximity to the inferior (nodosum) vagal ganglion (glomus vagale tumor, glomus intravagale tumor). Affected sites that are much less commonly reported are the periaortic area, trachea, larynx, mandible, nose, ciliary ganglion, and fallopian canal. [5]
In 1945, Rosenwasser described the first patient diagnosed with glomus jugulare tumor. [6] The patient survived until 1987. An association of glomus tumor with neurofibromatosis type 1 (NF-1) has been described. [7] Vascular tumors of the middle ear had previously been reported, but Rosenwasser was the first to recognize the origin of these tumors from the glomus jugulare. He provided the first description of the surgical removal of a glomus jugulare tumor.
Signs and symptoms
The most common symptoms are conductive hearing loss and pulsatile tinnitus. Other aural signs and symptoms are ear fullness, otorrhea, hemorrhage, bruit, and the presence of a middle ear mass. Significant ear pain is uncommon. Involvement of the inner ear produces vertigo and sensorineural hearing loss. [3]
Glomus jugulare tumors occur predominantly in women in the fifth and sixth decades of life. Because of the insidious onset of symptoms, these tumors often go unnoticed, and delay in diagnosis is frequent. Because of the location and extent of involvement, glomus jugulare tumors present significant diagnostic, management, and social challenges. [8, 9, 10, 11] Metastases from glomus tumors occur in approximately 4% of cases. [12, 13]
Examination
The most common finding during an otoscopic examination is a pulsatile, red middle-ear mass behind the tympanic membrane, which may show increased vascularity (a rising sun appearance). Sometimes, the tumor may erode into the ear canal, resulting in otorrhagia. [14, 3]
Audiologic examination reveals mixed conductive and sensorineural hearing loss. The sensorineural component tends to be more significant with larger tumors.
Plain skull radiography may show enlargement of the lateral jugular foramen and fossa. Axial and coronal computed tomography (CT) scanning with thin sections are superior at demonstrating the extent of bone destruction. Magnetic resonance imaging (MRI) with gadolinium-diethylenetriamine pentaacetic acid (DTPA) contrast is best for delineating tumor limits.
Treatment
Glomus jugulare tumors are challenging to treat because of their vascularization and location. [15] Treatment approaches remain controversial due to high morbidity. Historically, these tumors have primarily been managed surgically. [16] However, surgery may be contraindicated because of age or general physical condition. Surgical resection for type I tumors is relatively simple and complication free, but large tumors affecting the lower cranial nerves and extending beyond the petrous apex carry a significant risk of postoperative complications, especially in older patients. In such cases, embolization, radiation, Gamma Knife stereotactic radiosurgery, or intratumoral injection of cyanoacrylate glue may be considered. [1, 17, 18, 19, 20, 21, 22, 5]
(See the images below.)
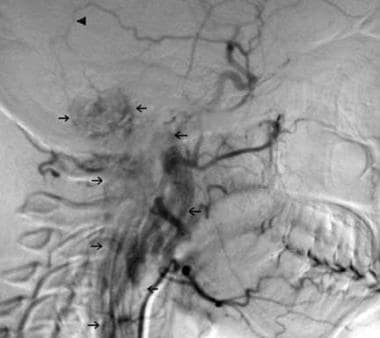 Lateral view of the initial carotid arteriogram of a 20-year-old woman who presented in June 1970 with episodic hypertension, headaches, and palpitations. Urine catecholamine levels were elevated, and a pheochromocytoma was suspected. She underwent a negative exploratory laparotomy. She subsequently developed palsies of the IX, X, XI, and XII cranial nerves on the right side. A norepinephrine-secreting glomus jugulare tumor with intracranial and cervical extension was identified on radiologic and arteriographic imaging. Arrows delineate the tumor blush. The arrowhead demonstrates a branch of the middle meningeal artery providing blood supply to the tumor. This branch was embolized.
Lateral view of the initial carotid arteriogram of a 20-year-old woman who presented in June 1970 with episodic hypertension, headaches, and palpitations. Urine catecholamine levels were elevated, and a pheochromocytoma was suspected. She underwent a negative exploratory laparotomy. She subsequently developed palsies of the IX, X, XI, and XII cranial nerves on the right side. A norepinephrine-secreting glomus jugulare tumor with intracranial and cervical extension was identified on radiologic and arteriographic imaging. Arrows delineate the tumor blush. The arrowhead demonstrates a branch of the middle meningeal artery providing blood supply to the tumor. This branch was embolized.
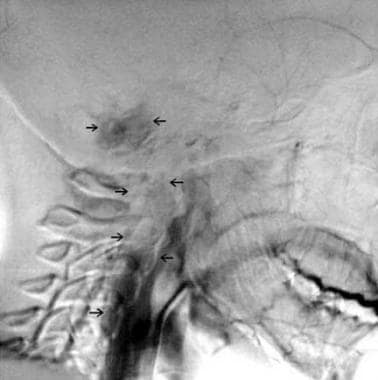 A significant decrease of tumor vascular blush (arrows) following embolization of a norepinephrine-secreting glomus jugulare tumor with intracranial and cervical extension.
A significant decrease of tumor vascular blush (arrows) following embolization of a norepinephrine-secreting glomus jugulare tumor with intracranial and cervical extension.
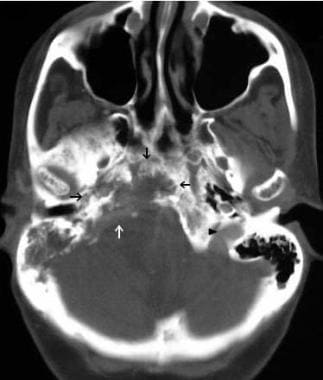 CT imaging demonstrates the extent of bony destruction (white and black arrows) by the tumor. The normal jugular foramen on the left (arrow head) is shown for comparison. The patient subsequently underwent surgical resection of the extracranial portion of this extensive tumor. The remaining intracranial portion was treated with radiation therapy (54 Gy). Follow-up evaluations, including imaging and laboratory investigations, demonstrated long-term control of both tumor growth and catecholamine production.
CT imaging demonstrates the extent of bony destruction (white and black arrows) by the tumor. The normal jugular foramen on the left (arrow head) is shown for comparison. The patient subsequently underwent surgical resection of the extracranial portion of this extensive tumor. The remaining intracranial portion was treated with radiation therapy (54 Gy). Follow-up evaluations, including imaging and laboratory investigations, demonstrated long-term control of both tumor growth and catecholamine production.
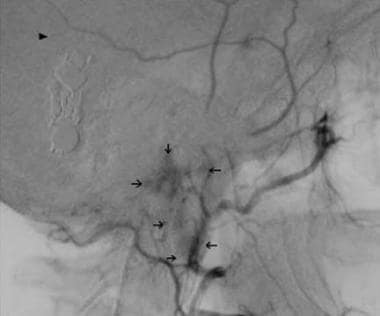 Lateral carotid arteriogram obtained 22 years after radiation therapy in a 20-year-old woman who presented in June 1970 with episodic hypertension, headaches, and palpitations.
Lateral carotid arteriogram obtained 22 years after radiation therapy in a 20-year-old woman who presented in June 1970 with episodic hypertension, headaches, and palpitations.
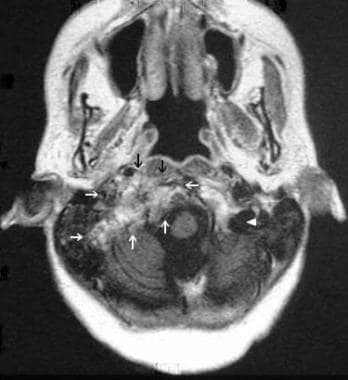 Corresponding MRI of the tumor depicted in the previous image indicating no evidence of tumor growth over time.
Corresponding MRI of the tumor depicted in the previous image indicating no evidence of tumor growth over time.
Glomus jugulare tumors are slow-growing tumors that require long-term follow-up by an interprofessional team, which should include primary care physicians, neurosurgeons, otolaryngologists, neuroradiologists, and physical medicine and rehabilitation teams. Patients and their families should be well educated about their disease, and the genetic screening of high-risk individuals in affected families has been advised to lower associated morbidity. [5]
Relevant Anatomy
Most jugulotympanic paraganglia are located in the adventitia of the jugular bulb within the jugular foramen. [2]
The main blood supply is via the ascending pharyngeal artery from the external carotid artery (ECA) and branches from the petrous portion of the internal carotid artery (ICA). Larger glomus jugulare tumors may also have blood supply from other branches of the ECA, ICA, vertebral artery, and thyrocervical trunk.
The walls of the jugular foramen are formed anterolaterally by the petrous bone and posteromedially by the occipital bone. The canal follows an anterior, inferior, and lateral direction to exit the skull.
The posterolateral portion of the foramen (pars venosa) contains the jugular bulb, posterior meningeal artery, and cranial nerves X and XI. The anteromedial portion (pars nervosa) contains the inferior petrosal sinus and cranial nerve IX. The jugular bulb is situated between the sigmoid sinus and the internal jugular vein. The lower cranial nerves are situated medial to the medial wall of the jugular bulb. The inferior petrosal sinus enters the medial aspect of the jugular bulb via several channels anterior to cranial nerves IX, X, and XI.
Many important structures are in proximity to the jugular bulb, including the internal auditory canal, the posterior semicircular canal, the middle ear, the medial external auditory canal, the facial nerve (posterolaterally), and the ICA (anteriorly) within the carotid canal. At the extracranial end of the jugular foramen, the ICA, internal jugular vein, and cranial nerves VII, X, XI, and XII are within a 2-cm area.
Pathophysiology
The glomera jugulare, or glomus bodies, are small collections of paraganglionic tissue. They are derived from embryonic neuroepithelium in close association with the autonomic nervous system and are found in the region of the jugular bulb. [23] Glomus tumors are encapsulated, slowly growing, highly vascular, and locally invasive tumors. Sen et al described the histologic structure of glomus tumors as a dense matrix of connective tissue among nerve fascicles. [24] These tumors tend to expand within the temporal bone via the pathways of least resistance, such as air cells, vascular lumens, skull base foramina, and the eustachian tube. They also invade and erode bone in a lobular fashion, but they often spare the ossicular chain.
Initially, the skull base erodes in the region of the jugular fossa and posteroinferior petrous bone, with subsequent extension to the mastoid and adjacent occipital bone (see the image below). Significant intracranial and extracranial extension may occur, as well as extension within the sigmoid and inferior petrosal sinuses. Neural infiltration is also common.
Lateral carotid arteriogram obtained 22 years after radiation therapy in a 20-year-old woman who presented in June 1970 with episodic hypertension, headaches, and palpitations.
The parenchyma of the paraganglia consists of 2 primary cell types. Type I cells are more common and are typically round with indistinct cell borders. Type II cells are smaller and irregularly shaped.
Metastases from glomus tumors occur in approximately 4% of cases. [12, 13] A reduction in the proportion of type II cells and a poorer staining of type I cells for s-100 and glial fibrillary acidic protein are reported to be correlated with an increased tumor grade. A metastatic lesion is distinguished from a multicentric lesion based on location. Metastases have been found in the lung, lymph nodes, liver, vertebrae, ribs, and spleen. Malignancy of the tumor probably is related to p53 and p16INK4A mutations.
Additional studies using immunohistochemical techniques revealed that malignant glomus tumors are characterized by the presence of MIB-1, p53, Bcl-2, and CD34. [25] Up to 4% of the tumors are functional and produce clinically significant levels of catecholamines, norepinephrine, or dopamine with symptoms mimicking a pheochromocytoma. Pheochromocytoma, parathyroid adenoma, and thyroid carcinoma have been reported in association with glomus jugulare tumors. [5]
The Glasscock-Jackson and Fisch classifications of glomus tumors are widely used. The Fisch classification of glomus tumors is based on extension of the tumor to surrounding anatomic structures and is closely related to mortality and morbidity. [26]
-
Type A tumor - Tumor limited to the middle ear cleft (glomus tympanicum)
-
Type B tumor - Tumor limited to the tympanomastoid area with no infralabyrinthine compartment involvement
-
Type C tumor - Tumor involving the infralabyrinthine compartment of the temporal bone and extending into the petrous apex
-
Type C1 tumor - Tumor with limited involvement of the vertical portion of the carotid canal
-
Type C2 tumor - Tumor invading the vertical portion of the carotid canal
-
Type C3 tumor - Tumor invasion of the horizontal portion of the carotid canal
-
Type D1 tumor - Tumor with an intracranial extension less than 2 cm in diameter
-
Type D2 tumor - Tumor with an intracranial extension greater than 2 cm in diameter
Etiology
Glomus jugulare tumors originate from the chief cells of the paraganglia, or glomus bodies, located within the wall (adventitia) of the jugular bulb, and can be associated with either the auricular branch of the vagus nerve (Arnold nerve) or the tympanic branch of the glossopharyngeal nerve (Jacobson nerve). Paraganglia are small (< 1.5 mm) masses of tissue composed of clusters of epithelioid (chief) cells within a network of capillary and precapillary caliber vessels. The number seems to increase until the fourth decade of life and then seems to decline. Paraganglia develop from the neural crest and are believed to function as chemoreceptors. Based on the presence of catecholamines and neuropeptides, paraganglia are included in the amine precursor uptake and decarboxylase (APUD) system, which has been referred to as the diffuse neuroendocrine system (DNES). [5]
Although most paragangliomas are sporadic, they can be familial with autosomal dominant inheritance and incomplete penetrance. The development of tumors in familial cases is dependent on age and on the sex of the affected parent. The nonchromaffin paragangliomas have a familial tendency. Tumors rarely occur in people younger than 18 years, and as a result of suspected genomic imprinting, only children of males possessing the disease gene develop tumors. The gene responsible for hereditary paragangliomas has been localized to band 11q23.
Epidemiology
Glomus tumors occur with an estimated annual incidence of 1 case per 1.3 million people. [27] Although rare, glomus tumors are the most common tumor of the middle ear and are second to vestibular schwannoma as the most common tumor of the temporal bone.
The female-to-male ratio is 3-6:1. Glomus jugulare tumors have also been noted to be more common on the left side, especially in females.
Most tumors occur in patients aged 40-70 years, but cases have been reported in patients as young as 6 months and as old as 88 years. Multicentric tumors are found in 3-10% of sporadic cases and in 25-50% of familial cases. [5]
Prognosis
Glomus jugulare tumors may grow slowly and produce cranial nerve palsies that, to a certain point, are benign and mostly cosmetic. However, despite this optimistic assessment, one study showed a long-term reduced quality of life in patients with glomus tumors. [8]
Prognosis has improved quite dramatically over the last decade, with a stroke rate of 0-3.5%, a cranial nerve injury rate of 5-39%, and overall mortality of 0-2.7%. With stereotactic radiosurgery treatment, 60% of the patients showed improvement of previous neurologic deficits, and tumor control was obtained in 91% of the patients. [28, 29]
Twenty years after treatment, the survival rate is 94%, and 77% of patients remain symptom free. In 1945, Rosenwasser described the first patient diagnosed with glomus jugulare tumor. The patient survived until 1987. [6]
Patient Education
Glomus jugulare are slow-growing tumors requireing long-term follow-up by an interprofessional team, which should include primary care physicians, neurosurgeons, otolaryngologists, and neuroradiologists as well as physical medicine and rehabilitation teams. Patients and their families should be well educated about their disease and the genetic screening of high-risk individuals in affected families has been advised to lower associated morbidity. [5]
-
Lateral view of the initial carotid arteriogram of a 20-year-old woman who presented in June 1970 with episodic hypertension, headaches, and palpitations. Urine catecholamine levels were elevated, and a pheochromocytoma was suspected. She underwent a negative exploratory laparotomy. She subsequently developed palsies of the IX, X, XI, and XII cranial nerves on the right side. A norepinephrine-secreting glomus jugulare tumor with intracranial and cervical extension was identified on radiologic and arteriographic imaging. Arrows delineate the tumor blush. The arrowhead demonstrates a branch of the middle meningeal artery providing blood supply to the tumor. This branch was embolized.
-
A significant decrease of tumor vascular blush (arrows) following embolization of a norepinephrine-secreting glomus jugulare tumor with intracranial and cervical extension.
-
CT imaging demonstrates the extent of bony destruction (white and black arrows) by the tumor. The normal jugular foramen on the left (arrow head) is shown for comparison. The patient subsequently underwent surgical resection of the extracranial portion of this extensive tumor. The remaining intracranial portion was treated with radiation therapy (54 Gy). Follow-up evaluations, including imaging and laboratory investigations, demonstrated long-term control of both tumor growth and catecholamine production.
-
Lateral carotid arteriogram obtained 22 years after radiation therapy in a 20-year-old woman who presented in June 1970 with episodic hypertension, headaches, and palpitations.
-
Corresponding MRI of the tumor depicted in the previous image indicating no evidence of tumor growth over time.



