Practice Essentials
Gigantism refers to abnormally high linear growth (see the image below) due to excessive action of insulinlike growth factor I (IGF-I) while the epiphyseal growth plates are open during childhood. Acromegaly is the same disorder of IGF-I excess but occurs after the growth plate cartilage fuses in adulthood.
 Gigantism and Acromegaly. Image shows a coauthor of this article with a statue of Robert Wadlow, who was called the Alton giant. The tallest person on record, he was 8 feet 11 inches tall at the time of his death.
Gigantism and Acromegaly. Image shows a coauthor of this article with a statue of Robert Wadlow, who was called the Alton giant. The tallest person on record, he was 8 feet 11 inches tall at the time of his death.
In acromegaly, a severe disease that is often diagnosed late, morbidity and mortality rates are high, particularly as a result of associated cardiovascular, cerebrovascular, and respiratory disorders and malignancies. [1]
Researchers have identified a gene on the X chromosome, GPR101, which was overexpressed 1000-fold more than normal in a genetic study of 43 patients affected by sporadic or inherited gigantism that manifested during childhood or adolescence. This duplication was not evident in patients who began abnormal growth at age 9 or 10, but only in those who started to grow excessively before the age of 3. In a separate analysis of 248 patients with sporadic acromegaly, a mutation in the GPR101 gene was found in about 4% of cases. [2, 3, 4] The GPR101 gene may be a target for the treatment of growth disorders.
A retrospective review of 208 cases of gigantism internationally established a genetic etiology in 46% of the cases; 29% had aryl hydrocarbon receptor interacting protein (AIP) gene mutations or deletions; and 10% had X-linked acro-gigantism (X-LAG) due to chromosome Xq26.3 microduplications on the GPR101 gene. The remaining 7% of genetic causes of gigantism were due to McCune-Albright syndrome (MAS), Carney complex, and MEN1. Male predominance was seen among AIP-mutated gigantism, whereas X-LAG had a strong female predilection. [5]
Signs and symptoms
Gigantism
The presentation of patients with gigantism is usually dramatic, unlike the insidious onset of acromegaly in adults. Manifestations include the following:
-
Tall stature
-
Mild to moderate obesity (common)
-
Macrocephaly (may precede linear growth)
-
Headaches
-
Visual changes
-
Hypopituitarism
-
Soft tissue hypertrophy
-
Exaggerated growth of the hands and feet, with thick fingers and toes
-
Coarse facial features
-
Frontal bossing
-
Prognathism
-
Hyperhidrosis
-
Osteoarthritis (a late feature of IGF-I excess)
-
Peripheral neuropathies (eg, carpel tunnel syndrome)
-
Cardiovascular disease
-
Benign tumors
-
Endocrinopathies
Acromegaly
Signs and symptoms of acromegaly include the following:
-
Doughy-feeling skin over the face and extremities
-
Thick and hard nails
-
Deepening of creases on the forehead and nasolabial folds
-
Noticeably large pores
-
Thick and edematous eyelids
-
Enlargement of the lower lip and nose (the nose takes on a triangular configuration)
-
Wide spacing of the teeth and prognathism
-
Cutis verticis gyrata (ie, furrows resembling gyri of the scalp) [6]
-
Small sessile and pedunculated fibromas (ie, skin tags)
-
Hypertrichosis
-
Oily skin (acne is not common)
-
Hyperpigmentation (40% of patients)
-
Acanthosis nigricans (a small percentage of patients)
-
Excessive eccrine and apocrine sweating
-
Breast tissue becoming atrophic; galactorrhea
-
High blood pressure
-
Mitral valvular regurgitation
-
Mild hirsutism (in women)
Diagnosis
Laboratory studies used in the diagnosis of growth hormone (GH)/IGF-I excess include the following:
-
Oral glucose: To determine the extent to which the patient can suppress GH concentration after the consumption of oral glucose
-
GH: Clearly elevated GH levels (>10 ng/mL) after oral glucose, combined with the clinical picture, secure the diagnosis of acromegaly
-
IGF-I: Elevated IGF-I values in a patient whose symptoms prompt appropriate clinical suspicion almost always indicate GH excess
Imaging studies include the following:
-
Magnetic resonance imaging (MRI): To image pituitary adenomas
-
Computed tomography (CT) scanning: To evaluate the patient for pancreatic, adrenal, and ovarian tumors secreting GH/GHRH; use chest CT scans to evaluate for bronchogenic carcinoma secreting GH/GHRH
-
Radiography: To demonstrate skeletal manifestations of GH/IGF-I excess
Management
No single treatment modality consistently achieves control of GH excess. For pituitary adenomas, transsphenoidal surgery is usually considered the first line of treatment, followed by medical therapy for residual disease. [7, 8] Radiation treatment usually is reserved for recalcitrant cases.
Somatostatin and dopamine analogues and GH receptor antagonists are the mainstays of medical treatment for GH excess and are generally used when primary surgery fails to induce complete remission.
Primary treatment with the somatostatin analogues depot octreotide and lanreotide has been found to induce tumor shrinkage in newly diagnosed acromegaly. [9] Dopamine-receptor agonists are generally used as adjuvant medical treatments for GH excess, and their effectiveness may be added to that of octreotide.
Radiation therapy is also generally recommended if GH hypersecretion is not normalized with surgery.
Background
Gigantism
Gigantism is a nonspecific term that refers to any standing height more than 2 standard deviations above the mean for the person's sex, age, and Tanner stage (ie, height Z score >+2). These disorders are placed along a spectrum of IGF-I hypersecretion, wherein the developmental stage when such excess originates determines the principal manifestations. The onset of IGF-I hypersecretion in childhood or late adolescence results in tall stature (see the image below). (See Presentation and Workup.)
 Gigantism and Acromegaly. Robert Wadlow, 19 years of age, with his father (postcard photo prior to 1937). Courtesy of Wikimedia Commons (https://commons.wikimedia.org/wiki/File:Robert_Wadlow_postcard.jpg).
Gigantism and Acromegaly. Robert Wadlow, 19 years of age, with his father (postcard photo prior to 1937). Courtesy of Wikimedia Commons (https://commons.wikimedia.org/wiki/File:Robert_Wadlow_postcard.jpg).
Scientific breakthroughs in the molecular, genetic, and hormonal basis of growth hormone (GH) excess have provided important insights into the pathogenesis, prognosis, and treatment of this exceedingly rare disease. (See Prognosis, Treatment, and Medication.)
Acromegaly
Acromegaly is a rare, insidious, and potentially life-threatening condition for which there is good, albeit incomplete, treatment that can give the patient additional years of high-quality life. [10] (See Prognosis, Treatment, and Medication.)
Symptoms develop insidiously, taking from years to decades to become apparent. The mean duration from symptom onset to diagnosis is 5-15 years, with a mean delay of 8.7 years. Excess GH produces a myriad of signs and symptoms and significantly increases morbidity and mortality rates. Additionally, the mass effect of the pituitary tumor itself can cause symptoms. Annual new patient incidence is estimated to be 3-4 cases per million population per year. The mean age at diagnosis is 40 years in males and 45 years in females. (See Presentation.)
Growth hormone and insulinlike growth factor
GH is necessary for normal linear growth. Its secretion from the pituitary gland is controlled by combined hypothalamic regulation, with secretion being stimulated by GHRH and inhibited by somatostatin (also called GH release–inhibiting hormone). Several tissues, including the endocrine pancreas, produce somatostatin in response to GH. (See Pathophysiology and Etiology.)
GH acts indirectly, by stimulating the formation of IGF hormones (also called somatomedins). IGF-I (somatomedin C), the most important IGF in postnatal growth, is produced in the liver, chondrocytes, kidneys, muscles, pituitary gland, and gastrointestinal tract.
Once released into the circulation, GH stimulates the production of IGF-I. The main source of circulating IGF-I is the liver, though it is produced in many other tissues. IGF-I is the primary mediator of the growth-promoting effects of GH.
It is characterized by increased and unregulated GH production, usually caused by a GH-secreting pituitary tumor (somatotroph tumor). Other causes of increased and unregulated GH production, all very rare, include increased GH-releasing hormone (GHRH) from hypothalamic tumors; ectopic GHRH from nonendocrine tumors; and ectopic GH secretion by nonendocrine tumors.
Pathophysiology and Etiology
Causes of excess IGF-I action can be divided into the following three categories:
-
Release of primary GH excess from the pituitary
-
Increased GHRH secretion or hypothalamic dysregulation
-
Hypothetically, the excessive production of IGF-binding protein, which prolongs the half-life of circulating IGF-I
By far, most people with gigantism or acromegaly have GH-secreting pituitary adenomas or hyperplasia. Other causes of increased and unregulated GH production, all very rare, include increased GHRH from hypothalamic tumors; ectopic GHRH from nonendocrine tumors; and ectopic GH secretion by nonendocrine tumors.
Although gigantism is typically an isolated disorder, rare cases occur as a feature of other conditions, such as the following:
Approximately 20% of patients with gigantism have McCune-Albright syndrome (the triad of precocious puberty, café au lait spots, fibrous dysplasia) and may have either pituitary hyperplasia or adenomas. (See the image below.)
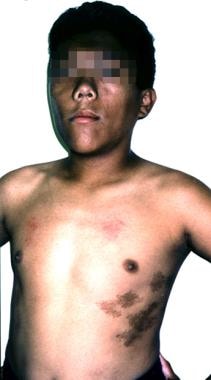 Gigantism and Acromegaly. A 12-year-old boy with McCune-Albright syndrome. His growth-hormone excess manifested as tall stature, coarse facial features, and macrocephaly.
Gigantism and Acromegaly. A 12-year-old boy with McCune-Albright syndrome. His growth-hormone excess manifested as tall stature, coarse facial features, and macrocephaly.
More than 95% of acromegaly cases are caused by a pituitary adenoma that secretes excess amounts of GH. Histopathologically, tumors include acidophil adenomas, densely granulated GH adenomas, sparsely granulated GH adenomas, somatomammotropic adenomas, and plurihormonal adenomas.
Ectopic production of GH and GHRH by malignant tumors accounts for other causes of IGF-I excess. (Ectopic GHRH-producing tumors, usually seen in the lung or pancreas, may occasionally be evident elsewhere, such as in the duodenum as a neuroendocrine carcinoma.) [12, 13]
Of these tumors, up to 40% have a mutation involving the alpha subunit of the stimulatory guanosine triphosphate (GTP)–binding protein. In the presence of a mutation, persistent elevation of cyclic adenosine monophosphate (cAMP) in the somatotrophs results in excessive GH secretion.
The pathologic effects of GH excess include acral overgrowth, insulin antagonism, nitrogen retention, increased risk of colon polyps/tumors, and acral overgrowth (ie, macrognathia; enlargement of the facial bone structure, as well as of the hands and feet; and visceral overgrowth, including macroglossia and enlargement of the heart muscle, thyroid, liver, and kidney). [14]
Pathologic studies on acromegalic hearts have shown extensive interstitial fibrosis, suggesting the existence of a specific acromegalic cardiomyopathy.
GH/IGF-I excess
Despite diverse pathophysiologic mechanisms, the final common abnormality in gigantism and acromegaly is IGF-I excess. Elevated tissue levels of free IGF-I, which is produced primarily in hepatocytes in response to excess GH, mediate most, if not all, growth-related outcomes in gigantism. Transgenic mice that overexpressed GH, GHRH, or IGF-I were found to have dramatically accelerated somatic growth compared with control litter mates.
One acromegalic patient had low serum GH levels and elevated serum total IGF-I levels; this finding implicates IGF-I as the key pathologic factor in this disease. Serum levels of IGF-I are consistently elevated in patients with acromegaly and, therefore, are used to monitor treatment success. The conditions described below can cause IGF-I oversecretion.
Primary pituitary GH excess
In most individuals with GH excess, the underlying anomaly is a benign pituitary tumor composed of somatotrophs (GH-secreting cells) or mammosomatotrophs (GH-secreting and prolactin-secreting cells) in the form of a pituitary microadenoma (< 1 cm) or macroadenoma (>1 cm). The adenomas are most characteristically well-demarcated and confined to the anterior lobe of the pituitary gland. In some people with GH excess, the tumor spreads outside the sella, invading the sphenoid bone, optic nerves, and brain. GH-secreting tumors are more likely to be locally invasive or aggressive in pediatric patients than in adults.
Gs-alpha (Gsa) mutation
G proteins play an integral role in postligand signal transduction in many endocrine cells by stimulating adenyl cyclase, resulting in an accumulation of cyclic adenosine monophosphate (cAMP) and subsequent gene transcription. About 20% of patients with gigantism have McCune-Albright syndrome and pituitary hyperplasia or adenomas.
Activating mutations of the stimulatory Gsa protein have been found in the pituitary lesions in McCune-Albright syndrome and are believed to cause the other glandular adenomas observed. Point mutations found in several tissues affected in McCune-Albright syndrome involve a single amino-acid substitution in codon 201 (exon 8) or 227 (exon 9) of the gene for Gsa. Somatic point mutations have been identified in the somatotrophs of less than 40% of sporadic GH-secreting pituitary adenomas. The resulting oncogene (gsp) is thought to induce tumorigenesis by persistently activating adenyl cyclase, with subsequent GH hypersecretion.
Loss of band 11q13 heterozygosity
Loss of heterozygosity at the site of a putative tumor-suppressor gene on band 11q13 was first identified in tumors from patients with MEN type I and GH excess. Loss of heterozygosity at band 11q13 has also been observed in all types of sporadically occurring pituitary adenomas. It is associated with an increased propensity for tumoral invasiveness and biologic activity.
Isolated familial somatotropinoma, a rare disease, refers to the occurrence of two or more cases of acromegaly or gigantism in a family in whom the features of Carney complex or MEN type 1 are absent. [15] It appears to be inherited as an autosomal dominant disease with incomplete penetrance. Although an association exists between isolated familial somatotropinoma and loss of heterozygosity on 11q13, the responsible gene remains unknown.
Abnormality at Carney loci on chromosomes 2 and 17
The Carney complex, which is characterized by myxomas, endocrine tumors, and spotty pigmentation, is transmitted as an autosomal dominant trait. About 8% of affected individuals have GH-producing pituitary adenomas. The causative gene for this disease was mapped to bands 2p16 and 17q22-24. Germline mutations in PRKAR1A (which encodes for the protein kinase A type I-alpha regulatory subunit, an apparent tumor-suppressor gene on chromosome arm 17q) were detected in several families with Carney complex.
Secondary GH excess
Causes of secondary GH excess include increased secretion of GHRH due to an intracranial or ectopic source and dysregulation of the hypothalamic-pituitary-GH axis.
GHRH excess
Hypothalamic GHRH excess is postulated as a cause for gigantism, possibly secondary to an activating mutation in hypothalamic GHRH neurons. Excess GHRH secretion may be due to an intracranial or ectopic tumor. Several well-documented incidents of hypothalamic GHRH excess demonstrated intracranial gangliocytomas associated with gigantism or acromegaly.
Ectopic GHRH-secreting tumors have included carcinoid, pancreatic islet-cell, and bronchial neoplasms. Prolonged tumoral secretion of GHRH leads to pituitary hyperplasia, with or without adenomatous transformation, that increases levels of GH and other adenohypophyseal peptides.
Disruption of somatostatin tone
Tumoral infiltration into somatostatinergic pathways are hypothesized to be the basis for GH excess in rare incidents of gigantism associated with neurofibromatosis and optic glioma or astrocytomas.
Epidemiology
Gigantism is extremely rare, with approximately 100 reported cases to date. Although still rare, acromegaly is more common than gigantism, with a prevalence of 36-69 cases per million and an incidence of 3-4 cases per million per year. [16]
Gigantism may begin at any age before epiphyseal fusion. X-linked acrogigantism (X-LAG) caused by microduplications on chromosome Xq26.3, encompassing the gene GPR101, is a severe infant-onset gigantism syndrome with onset as early as 2-3 months of age (median, 12 months). [4] Other genetic causes of gigantism include familial isolated pituitary adenoma (FIPA) caused by aryl hydrocarbon receptor interacting protein (AIP) gene mutations, multiple endocrine neoplasia type 1 (MEN1), McCune-Albright syndrome (MAS), and Carney complex with onset during prepubescence. [5]
The mean age for onset of acromegaly is in the third decade of life; the delay from the insidious onset of symptoms to diagnosis is 5-15 years, with a mean delay of 8.7 years. The mean age at diagnosis for acromegaly is 40 years in males and 45 years in females.
Prognosis
Because of the small number of people with gigantism, mortality and morbidity rates for this disease during childhood are unknown.
In acromegaly, a severe disease that is often diagnosed late, morbidity and mortality rates are high, particularly as a result of associated cardiovascular, cerebrovascular, and respiratory disorders and malignancies. [1]
Because IGF-I is a general growth factor, somatic hypertrophy in acromegaly occurs across all organ systems. Associated complications include the following [17] :
-
Acromegalic heart
-
Increased muscle and soft tissue mass
-
Increased kidney size
-
Articular overgrowth of synovial tissue and hypertrophic arthropathy
-
Joint symptoms, back pain, and kyphosis: Common presenting features
-
Thick skin
-
Hyperhidrosis (often malodorous)
-
Carpal tunnel syndrome and other entrapment syndromes
-
Macroglossia: May result in sleep apnea
-
Cerebral aneurysm and increased risk of cerebrovascular accident: Less common [18]
The prevalence of gastritis, duodenitis, peptic ulcer and intestinal metaplasia may be enhanced in acromegaly as compared to a normal healthy population. [19]
Early diagnosis of acromegaly, however, results in early transsphenoidal pituitary microsurgery, and currently, patients are more likely to be cured than in the past.
Reversal of excessive GH produces the following:
-
Decreased soft tissue swelling
-
Diminished sweating
-
Restoration of normal glucose tolerance
No studies have established, however, that the treatment of acromegaly leads to a reduction in morbidity and mortality rates, although successful treatment, with normalization of IGF-I levels, may be associated with a return to normal life expectancy.
Remission depends on the initial size of the tumor, the patient’s GH level, and the skill of the neurosurgeon. Remission rates of 80-85% and 50-65% can be expected for microadenomas and macroadenomas, respectively.
The postoperative GH concentration may predict remission rates. According to the results of one study, a postoperative GH concentration of less than 3 ng/dL was associated with a 90% remission rate, which declined to 5% in patients with a postoperative GH concentration of greater than 5 ng/dL.
Metabolic and endocrine complications
Diabetes mellitus occurs in 10-20% of patients with acromegaly. A 2009 study suggests that in patients with acromegaly, insulin resistance and hyperinsulinemia are positively correlated with the level of disease activity. [20] Hypertriglyceridemia is found in 19-44% of patients. Multinodular goiter also is often present in acromegaly.
Hypopituitarism may develop in patients with acromegaly, as a result of the pituitary mass or as a complication of surgery or radiation therapy. Treat pituitary failure with appropriate hormone-replacement therapy.
Respiratory complications
In acromegaly, respiratory complications occur as follows:
-
Increased lung capacity: 81% of men and 56% of women
-
Small airway narrowing: 36% of patients
-
Upper airway narrowing: 26% of patients
-
Acute dyspnea and stridor
-
Sleep apnea: As a significant cause of morbidity, sleep apnea may be both obstructive and central; curing acromegaly does not necessarily correct the disorder
Cardiovascular complications
A study by Berg et al found an increased prevalence of cardiovascular risk factors in patients with acromegaly compared with controls. [1] Cardiovascular complications include the following:
-
Hypertension
-
Acromegalic cardiomyopathy (with dysfunction and arrhythmias)
-
Left ventricular hypertrophy
-
Increased left ventricular mass
Disorders of calcium and bone metabolism
The following calcium and bone metabolism disorders can be found in acromegaly:
-
Hypercalciuria
-
Hyperphosphatemia
-
Urolithiasis
Neuromuscular complications
In acromegaly, these include the following:
-
Weakness (although with muscular appearance)
-
Nerve root compression
-
Radiculopathy
-
Spinal stenosis
-
Carpal tunnel syndrome
Cancer risks
Patients with acromegaly may be at increased risk for colorectal cancer and premalignant adenomatous polyps. Most studies suggest that as many as 30% of patients may have a premalignant colon polyp at diagnosis and that as many as 5% may have a colonic malignancy. [21] In studies, polyps were generally multiple and proximal to the splenic flexure, making them less likely to be discovered during sigmoidoscopy. However, the long-term effect of colonic lesions on morbidity and mortality has not been established.
Patients with acromegaly may also have an increased risk of developing breast and prostate tumors, although no clear evidence supports this; the risk of thyroid cancer is increased in males. [14] However, the prevalence of cancers in patients with acromegaly remains controversial, although patients might be advised to undergo screening colonoscopy and thyroid ultrasonography. [21, 22, 23]
Mortality
For individuals with acromegaly, the mortality rate is 2-3 times that of the general population, with cardiovascular and respiratory complications being the most frequent causes of death. Transgenic mouse models of acromegaly demonstrate cardiac and vascular hypertrophy but normal function, raising the concern that hypertrophic cardiomyopathy may contribute to the increased mortality. [24]
A study by Bates et al suggested that the extent of a patient’s GH excess impacts mortality. The investigators found that acromegaly patients with a GH concentration of greater than 10 ng/mL had double the expected mortality rate, whereas patients with a GH concentration of less than 5 ng/mL approached normal mortality. [16] These results underscore the necessity to reduce GH and IGF-I concentration in patients with acromegaly.
Researchers disagree on whether malignancy is a significant cause of increased mortality in acromegaly. Although benign tumors (including uterine myomas, prostatic hypertrophy, and skin tags) are frequently encountered in acromegaly, documentation for overall prevalence of malignancies in patients with acromegaly remains controversial.
Patient Education
Refer patients to the Hormone Health Network for additional information.
For patient education information, the Thyroid & Metabolism Center, as well as, Acromegaly, Acromegaly FAQs, and Acromegaly Medications.
-
Gigantism and Acromegaly. Image shows a coauthor of this article with a statue of Robert Wadlow, who was called the Alton giant. The tallest person on record, he was 8 feet 11 inches tall at the time of his death.
-
Gigantism and Acromegaly. A 12-year-old boy with McCune-Albright syndrome. His growth-hormone excess manifested as tall stature, coarse facial features, and macrocephaly.
-
Gigantism and Acromegaly. Robert Wadlow, 19 years of age, with his father (postcard photo prior to 1937). Courtesy of Wikimedia Commons (https://commons.wikimedia.org/wiki/File:Robert_Wadlow_postcard.jpg).





