Practice Essentials
Bronchiectasis is an uncommon disease, most often secondary to an infectious process, that results in the abnormal and permanent distortion of one or more of the conducting bronchi or airways.
In 1950, Reid characterized bronchiectasis as cylindrical, cystic, or varicose in nature. [1] Cylindrical bronchiectasis involves diffuse mucosal edema, with resultant bronchi that are dilated but have straight, regular outlines that end squarely and abruptly (see the image below).
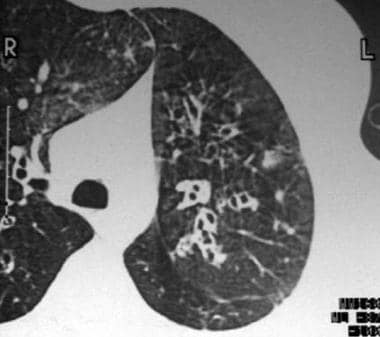 Cylindrical bronchiectasis with signet-ring appearance. Note that the luminal airway diameter is greater than the diameter of the adjacent vessel.
Cylindrical bronchiectasis with signet-ring appearance. Note that the luminal airway diameter is greater than the diameter of the adjacent vessel.
Signs and symptoms
Clinical manifestations of bronchiectasis are as follows:
-
Cough and daily mucopurulent sputum production, often lasting months to years (classic)
-
Blood-streaked sputum or hemoptysis from airway damage associated with acute infection
-
Dyspnea, pleuritic chest pain, wheezing, fever, weakness, fatigue, and weight loss
-
Rarely, episodic hemoptysis with little to no sputum production (ie, dry bronchiectasis)
Exacerbations of bronchiectasis from acute bacterial infections may produce the following signs:
-
Increased sputum production over baseline
-
Increased viscidity of sputum
-
A foul odor of the sputum (occasional)
-
Low-grade fever (rare)
-
Increased constitutional symptoms (eg, fatigue, malaise)
-
Increased dyspnea, shortness of breath, wheezing, or pleuritic pain
Findings on physical examination are nonspecific and may include the following:
-
Crackles, rhonchi, scattered wheezing, and inspiratory squeaks on auscultation
-
Digital clubbing (2-3% of patients; more frequent in moderate-to-severe cases)
-
Cyanosis and plethora with polycythemia from chronic hypoxia (rare)
-
Wasting and weight loss
-
Nasal polyps and signs of chronic sinusitis
-
Physical stigmata of cor pulmonale, in advanced disease
See Clinical Presentation for more detail.
Diagnosis
The diagnosis of bronchiectasis involves the following:
-
A compatible history of chronic respiratory symptoms (eg, daily cough and purulent sputum production)
-
Sputum analysis may strengthen clinical suspicion
-
Chest radiography is occasionally sufficient for confirming the diagnosis
Tests to identify underlying illnesses include the following:
-
Quantitative immunoglobulin levels, to exclude hypogammaglobulinemia
-
Quantitative serum alpha1-antitrypsin (AAT) levels, to rule out AAT deficiency
-
Aspergillus precipitins and serum total IgE levels, to diagnose ABPA
-
Autoimmune screening tests
Vitamin D deficiency is common in bronchiectasis and correlates with markers of disease severity. Chalmers et al measured serum 25-hydroxyvitamin-D by immunoassay in 402 stable patients with bronchiectasis and found that 50% were vitamin D deficient (levels below 25 nmol/L), compared with only 12% of matched controls, and 43% were vitamin D insufficient (25-74 nmol/L). Vitamin D–deficient bronchiectasis patients were more likely to be colonized with Pseudomonas aeruginosa, had lower forced expiratory volume in 1 second (FEV1) percent predicted, and had more frequent pulmonary exacerbations. [5]
Pulmonary function test results may be normal or abnormal; abnormalities are as follows:
-
Abnormalities may reflect underlying comorbidities and predisposing conditions
-
The most common abnormality is an obstructive airway defect, which is not usually reversible with bronchodilator therapy
-
A subgroup of patients have hyperreactive airways that will respond to bronchodilators
-
Yearly decline in FEV1 is greater in patients with bronchiectasis
Expected general findings on posterior-anterior and lateral chest radiographs include the following:
-
Increased pulmonary markings
-
Honeycombing
-
Atelectasis
-
Pleural changes
Specific findings on chest radiographs may include the following:
-
Linear lucencies and parallel markings radiating from the hila (tram tracking) in cylindrical bronchiectasis
-
Dilated bronchi in varicose bronchiectasis
-
Clustered cysts in cystic bronchiectasis
Noteworthy CT findings in bronchiectasis include the following:
-
Cylindrical bronchiectasis has parallel tram track lines, or it may have a signet-ring appearance composed of a dilated bronchus cut in a horizontal section with an adjacent pulmonary artery representing the stone
-
The diameter of the bronchus lumen is normally 1-1.5 times that of the adjacent vessel; a diameter greater than 1.5 times that of the adjacent vessel suggests bronchiectasis
-
Varicose bronchiectasis has irregular or beaded bronchi, with alternating areas of dilatation and constriction
-
Cystic bronchiectasis has large cystic spaces and a honeycomb appearance; this contrasts with the blebs of emphysema, which have thinner walls and are not accompanied by proximal airway abnormalities
See Workup for more detail.
Management
Treatment modalities include the following:
-
Antibiotics and chest physiotherapy are the mainstays
-
Bronchodilators
-
Corticosteroid therapy
-
Dietary supplementation
-
Oxygen (reserved for hypoxemic patients with severe disease)
-
Hospitalization for severe exacerbations
-
Surgical therapies
Acceptable antibiotic regimens for mild to moderately ill outpatients include 7-10 days of any of the following:
-
Amoxicillin
-
Tetracycline
-
Trimethoprim-sulfamethoxazole
-
A second-generation cephalosporin
-
A fluoroquinolone
For patients with moderate-to-severe symptoms, parenteral administration of the following antibiotics may be indicated:
-
An aminoglycoside (eg, gentamicin, tobramycin) and
-
An antipseudomonal synthetic penicillin, a third-generation cephalosporin, or a fluoroquinolone
-
Tobramycin, for patients infected with mucoid Pseudomonas species
American Thoracic Society recommendations for treatment of Mycobacterium avium complex (MAC) infection in the setting of bronchiectasis are as follows:
-
Combination therapy with clarithromycin, rifampin, ethambutol
-
Consider streptomycin as a possible fourth drug
-
Continue therapy until the patient's culture results have been negative for 1 year
-
The typical duration of therapy is 18-24 months
Surgical resection of involved bronchiectatic sites is an important adjunct to therapy for patients with focal disease that is poorly controlled by antibiotics. Other indications for surgical intervention may include the following:
-
Reduction of acute infective episodes
-
Reduction of excessive sputum production
-
Massive hemoptysis (alternatively, bronchial artery embolization may be attempted)
-
Foreign body or tumor removal
-
Consideration in the treatment of MAC or Aspergillus species infections
See Treatment and Medication for more detail.
Background
Bronchiectasis is an uncommon disease, most often secondary to an infectious process, that results in the abnormal and permanent distortion of one or more of the conducting bronchi or airways. First described by Laennec in 1819, later detailed by Sir William Osler in the late 1800s, and further defined by Reid in the 1950s, bronchiectasis has undergone significant changes in regard to its prevalence, etiology, presentation, and treatment. [9]
Bronchiectasis can be categorized as a chronic obstructive pulmonary disease manifested by airways that are inflamed and easily collapsible, resulting in air flow obstruction with shortness of breath, impaired clearance of secretions (often with disabling cough), and occasionally hemoptysis. Severe cases can result in progressive impairment with respiratory failure. [10, 11]
Bronchiectasis most commonly presents as a focal process involving a lobe, segment, or subsegment of the lung. Far less commonly, it may be a diffuse process involving both lungs; these cases most often occur in association with systemic illnesses, such as cystic fibrosis (CF), sinopulmonary disease, or both. The majority of this article will address non-CF related bronchiectasis.
Diagnosis is usually based on a compatible clinical history of chronic respiratory symptoms, such as a daily cough and viscid sputum production (see Clinical), and characteristic radiographic findings on CT scans, such as bronchial wall thickening and luminal dilatation (see Workup).
Antibiotics and chest physiotherapy are the mainstay modalities. Additionally, management of underlying conditions, such as hypogammaglobulinemia or alpha1-antitrypsin deficiency, is essential to the overall treatment. Surgery is an important adjunct to therapy in some patients with advanced or complicated disease. (See Treatment.)
For a discussion of this disorder in children, see the article Pediatric Bronchiectasis.
Pathophysiology
Bronchiectasis is an abnormal dilation of the proximal and medium-sized bronchi (>2 mm in diameter) caused by weakening or destruction of the muscular and elastic components of the bronchial walls. Affected areas may show a variety of changes, including transmural inflammation, edema, scarring, and ulceration, among other findings. Distal lung parenchyma may also be damaged secondary to persistent microbial infection and frequent postobstructive pneumonia. Bronchiectasis can be congenital but is most often acquired. [9]
Congenital bronchiectasis usually affects infants and children. These cases result from developmental arrest of the bronchial tree.
Acquired forms occur in adults and older children and require an infectious insult, impairment of drainage, airway obstruction, and/or a defect in host defense. The tissue is also damaged in part by the host response of neutrophilic proteases, inflammatory cytokines, nitric oxide, and oxygen radicals. This results in damage to the muscular and elastic components of the bronchial wall. Additionally, peribronchial alveolar tissue may be damaged, resulting in diffuse peribronchial fibrosis. [12]
The result is abnormal bronchial dilatation with bronchial wall destruction and transmural inflammation. The most important functional finding of altered airway anatomy is severely impaired clearance of secretions from the bronchial tree.
Impaired clearance of secretions causes colonization and infection with pathogenic organisms, contributing to the purulent expectoration commonly observed in patients with bronchiectasis. The result is further bronchial damage and a vicious cycle of bronchial damage, bronchial dilation, impaired clearance of secretions, recurrent infection, and more bronchial damage. [13]
In 1950, Reid characterized bronchiectasis as cylindrical, cystic, or varicose in nature. [1] Cylindrical bronchiectasis involves diffuse mucosal edema, with resultant bronchi that are dilated but have straight, regular outlines that end squarely and abruptly (see the image below).
Cylindrical bronchiectasis with signet-ring appearance. Note that the luminal airway diameter is greater than the diameter of the adjacent vessel.
Cystic or saccular bronchiectasis has ulceration with bronchial neovascularization. The result is a ballooned appearance and sometimes air-fluid levels (see the image below).
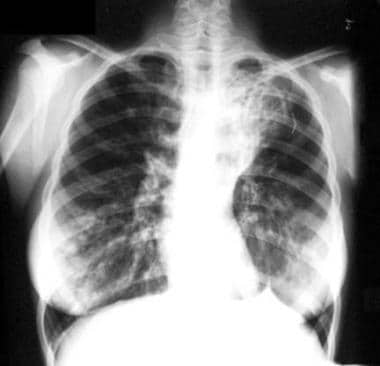 Cystic and cylindrical bronchiectasis of the right lower lobe on a posterior-anterior chest radiograph.
Cystic and cylindrical bronchiectasis of the right lower lobe on a posterior-anterior chest radiograph.
Varicose bronchiectasis has a bulbous appearance with a dilated bronchus and interspersed sites of relative constriction and, potentially, obstructive scarring. The latter may subsequently result in postobstructive pneumonitis and additional parenchymal damage (see the image below).
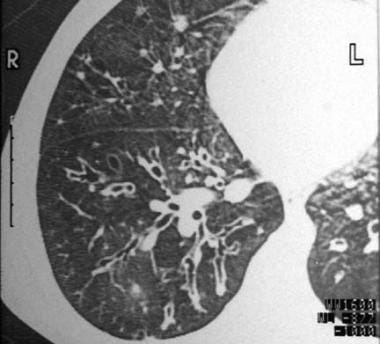 Varicose bronchiectasis with alternating areas of bronchial dilatation and constriction.
Varicose bronchiectasis with alternating areas of bronchial dilatation and constriction.
Etiology
Causes of bronchiectasis include the following:
-
Primary infections
-
Bronchial obstruction
-
Aspiration
-
Primary ciliary dyskinesia
-
Allergic bronchopulmonary aspergillosis
-
Immunodeficiency states
-
Congenital anatomic defects
-
Connective-tissue disorders
-
Autoimmune diseases
-
Idiopathic inflammatory disorders
-
Autosomal dominant polycystic kidney disease
-
Traction from other processes
-
Toxic gas exposure
Primary infections
Bronchiectasis may be the sequela of a variety of necrotizing infections that are either inadequately treated or not treated at all. Primary infection (ie, in the absence of intrinsic defects or noninfectious extrinsic insults) was a particularly common cause of bronchiectasis in developed countries prior to the widespread use of antibiotics [14] and it remains important in developing countries, where antibiotics are used inconsistently. [15, 16]
Typical offending organisms that have been known to cause bronchiectasis include the following [17, 14] :
-
Klebsiella species
-
Staphylococcus aureus
-
Mycobacterium tuberculosis
-
Mycoplasma pneumoniae
-
Nontuberculous mycobacteria
-
Measles virus
-
Pertussis virus
-
Influenza virus
-
Herpes simplex virus
-
Certain types of adenovirus
Infection with respiratory syncytial virus in childhood may also result in bronchiectasis.
Mycobacterium avium complex (MAC) infection deserves special mention. It has a propensity to occur in the setting of human immunodeficiency virus (HIV) infection as well as in hosts who are immunocompetent. [18]
MAC infection has been observed especially in women who are nonsmokers; are older than 60 years; do not have a known predisposing pulmonary disorder; and tend to voluntarily suppress cough. [19] Sputum smear in these cases is positive for acid-fast bacilli, and CT scan shows small regular nodules and findings of bronchiectasis. [20, 21, 19]
Once a patient develops bronchiectasis, many of these same organisms colonize the damaged bronchi and may cause ongoing damage and episodic infectious exacerbations. The organisms found most typically include Haemophilus species (47-55% of patients) and Pseudomonas species (18-26% of patients). [22, 23]
Although not a primary cause of bronchiectasis, P aeruginosa often causes chronic bronchial infection in patients with non-CF bronchiectasis via a mechanism involving biofilm formation and the release of virulence factors. This suggests that Pseudomonas species may promote disease progression, and that infection with these species may be related to worsening lung function and increased morbidity and mortality. [24]
Bronchial obstruction
Focal postobstructive bronchiectasis may occur in a number of clinical settings (eg, endobronchial tumors, broncholithiasis, bronchial stenosis from infections, encroachment of hilar lymph nodes, foreign body aspiration). Right-middle lobe syndrome is a specific type of bronchial obstruction that may result in bronchiectasis. It results from an abnormal angulation of the lobar bronchus at its origin, predisposing it to obstruction, subsequent infection, and development of bronchiectasis.
Aspiration
In adults, foreign body aspiration often takes place in the setting of altered mental status and involves unchewed food. Patients may also aspirate chewed materials from the stomach, including food, peptic acid, and microorganisms.
After aspiration, a postobstructive pneumonia may occur, with subsequent development of focal bronchiectasis. Bronchiectasis may also develop in the setting of chronic aspiration. Further recognized is that a history of gastroesophageal reflux is a risk factor for aspiration and that the organism Helicobacter pylori may play a role in the development of bronchiectasis in this group of patients. [25, 26, 27]
Cystic fibrosis
CF is a multisystem disorder that affects the chloride transport system in exocrine tissues, primarily secondary to a defect in the CF transmembrane regulator (CFTR) protein. CF and its variants are the most common cause of bronchiectasis in the United States and other industrialized nations.
CF is an autosomal recessive disease affecting approximately 1 in 2,500 whites and 1 in 17,000 blacks in the United States. [28] In was estimated that in 2005, 10,000 adults in the United States would have CF, comprising 40% of the total CF population. [29]
Multiple genetic variants of CF exist, and the risk to patients that have genetic heterozygous mutations remains to be elucidated. However, a reasonable assumption is that patients with CF can be divided into 2 groups: (1) those with classic disease that is readily diagnosed based on clinical and laboratory data and (2) those with less severe disease that manifests later in life and who have ambiguous genetic testing results. [30, 31, 32]
The major pulmonary finding in CF is bronchiectasis, which is an almost universal feature of this disease. It may be the sole feature of CF in adults or those with genetic variations of the disease. Bronchiectasis associated with CF is believed to occur secondary to mucous plugging of proximal airways and chronic pulmonary infection, especially with mucoid P aeruginosa. [33]
Young syndrome
Young syndrome is clinically similar to CF and may represent a genetic variant of CF. It is most often observed in middle-aged men in North America and is a leading cause of male infertility. [34]
Patients with Young syndrome have bronchiectasis (often predominant in the lower lobes), sinusitis, and obstructive azoospermia. However, they do not display the other findings of CF. The pathogenesis of bronchiectasis in these patients is believed to be similar to that of bronchiectasis in CF. The criterion standard for diagnosis of Young syndrome is electron microscopic analysis of the structure of the cilia.
Primary ciliary dyskinesia
Primary ciliary dyskinesia is a group of inherited disorders that may affect 1 in 15,000-30,000 population. It is manifested by immotile or dyskinetic cilia and/or sperm. This may lead to poor mucociliary clearance, recurrent pulmonary infections, and, ultimately, bronchiectasis. [35, 36]
A variant of this condition, initially described by Kartagener, encompassed the clinical triad of situs inversus, nasal polyps or sinusitis, and bronchiectasis in the setting of immotile cilia of the respiratory tract. [37]
Allergic bronchopulmonary aspergillosis
Allergic bronchopulmonary aspergillosis (ABPA) is a hypersensitivity reaction to inhaled Aspergillus antigen that is characterized by bronchospasm, bronchiectasis, and immunologic evidence of a reaction to Aspergillus species. [38] ABPA should be suspected in patients with a productive cough who also have a long history of asthma-type symptoms that do not respond to conventional therapy.
Bronchiectasis is believed to be secondary to airway plugging by viscid secretions containing hyphae of Aspergillus species. The resulting bronchiectasis is thin-walled and affects the central and medium-sized airways.
CT scanning of the chest demonstrates central airway bronchiectasis, differentiating this condition from other causes of bronchiectasis. Other features of ABPA include eosinophilia, elevated immunoglobulin E (IgE) levels, and dramatic responses to therapeutic corticosteroids.
Immunodeficiency states
Immunodeficiency states may be congenital or acquired. The most common congenital conditions (albeit rare) involve B-lymphocyte functions. Hypogammaglobulinemia in these cases may take one of the following forms [39, 40, 41, 42] :
-
Immunoglobulin G (IgG) subclass deficiency
-
X-linked agammaglobulinemia
-
Immunoglobulin A (IgA) deficiency
-
Immunoglobulin M (IgM) deficiency
-
Immunoglobulin E (IgE) deficiency
Patients with hypogammaglobulinemia usually present in childhood with repeated sinus or pulmonary infections, although the disorder has been diagnosed in adults who did not have a history of repeated infections. Establishing the diagnosis is important because gammaglobulin replacement may reduce the number of infections and resultant lung injury.
HIV disease, with resultant acquired immunodeficiency syndrome (AIDS), has been implicated in the development of bronchiectasis and demonstrates the accelerated bronchial damage that may occur from repeated infections in patients who are immunosuppressed. Bronchiectasis in HIV infection has occurred with and without obvious preceding pulmonary infection and may occur secondary to immunologic dysfunction from the HIV disease itself. [18, 43, 44]
Congenital anatomic defects
Bronchiectasis can result from a variety of congenital anatomic defects. Bronchopulmonary sequestration is a congenital abnormality classified as either intralobar or extralobar and results in chronic lower respiratory tract infections that lead to bronchiectasis.
Williams-Campbell syndrome (congenital cartilage deficiency) is the absence of cartilage from lobar to first- to second-generation segmental airways that results in extensive peripheral bronchiectasis. [45]
Mounier-Kuhn syndrome (tracheobronchomegaly) is a rare disorder characterized by dilation of the trachea and segmental bronchi (central bronchiectasis). [46]
Swyer-James syndrome (unilateral hyperlucent lung) likely is a developmental disturbance that leads to unilateral bronchiolitis, hyperinflation, and, in some cases, bronchiectasis.
Yellow-nail syndrome is rare. It results in exudative pleural effusions. [47]
Alpha1-antitrypsin (AAT) deficiency
Bronchiectasis has been noted to occur in this rare condition, both in patients with true AAT deficiency and in patients with heterozygous phenotypes. [48, 49, 50, 51]
The pathogenesis of bronchiectasis in this setting is unclear, but it is believed that the AAT abnormalities make patients more susceptible to respiratory tract infections and subsequent bronchial damage.
Autoimmune diseases, connective-tissue disorders, and idiopathic inflammatory disorders
Rheumatoid arthritis is associated with bronchiectasis in a reported 3.2-35% of patients [52, 53, 54] and, in one series, was associated with an unfavorable prognosis. [55] The pathology of bronchiectasis may be increased susceptibility to infections in these patients. Pulmonary disease may occur prior to the onset of the rheumatic process.
Bronchiectasis has been noted in patients with Sjögren syndrome and may be secondary to increased viscosity of mucus with poor airway clearance. [56]
Ankylosing spondylitis is associated with bronchiectasis, but in small numbers. [57]
Systematic lupus erythematosus may present with a variety of pulmonary pathology, including bronchiectasis, which was reported in 21% of patients in one series. [58]
In relapsing polychondritis, bronchiectasis appears to be secondary to primary bronchial damage with resultant recurrent infection. [59]
With inflammatory bowel disease, bronchiectasis has been seen in both ulcerative colitis and Crohn disease. The etiology remains unclear. Pulmonary symptoms may occur prior to the onset of bowel disease. [60]
Sarcoidosis may cause bronchiectasis by a variety of mechanisms, including parenchymal scarring, endobronchial granulomatous inflammation, or extrinsic compression of bronchi. [61]
Marfan syndrome is a connective tissue disorder. The general consensus is that weakness of the connective tissue of the bronchial wall predisposes to bronchiectasis. [62]
Autosomal dominant polycystic kidney disease
Autosomal dominant polycystic kidney disease (ADPKD) patients have also been shown to have an increased incidence of bronchiectasis on radiographic screening. ADPKD is another of the so-called "ciliopathies," or diseases in which a defect in ciliary function is the primary pathologic finding. [63]
Traction bronchiectasis
Traction bronchiectasis is distortion of the airways secondary to mechanical traction on the bronchi from fibrosis of the surrounding lung parenchyma. Although the airways may become dilated in this situation, the other manifestations of bronchiectasis are lacking. Traction bronchiectasis tends to have an upper lobe distribution in cases of radiation fibrosis and sarcoidosis, while the lower lobe is predominantly involved in cases of interstitial lung disease/ idiopathic pulmonary fibrosis (ILD/IPF). [64]
Toxic gas exposure
Exposure to toxic gas may often cause irreversible damage to the bronchial airways and cystic bronchiectasis. Commonly implicated agents include chlorine gas and ammonia.
Epidemiology
Currently no systematic data are available on the incidence or prevalence of bronchiectasis. A general theory is that the emergence of vaccines and antibiotics in the 20th century resulted in a decline in the rate of bronchiectasis in developed countries. [17]
The best data available suggest that the prevalence of bronchiectasis mirrors the socioeconomic conditions of the population under study, with significantly lower prevalence in areas where immunizations and antibiotics are readily available. Bronchiectasis remains a major cause of morbidity in less-developed countries, especially in countries with limited access to medical care and antibiotic therapy. [15, 16]
United States statistics
Bronchiectasis is relatively uncommon in the United States, with a prevalence of approximately 100,000 cases, based on data from the 1980s. That said, the number of bronchiectasis cases in the United States associated with atypical mycobacteria or other environmental factors reportedly has increased, [20, 21, 65] perhaps due to improved detection techniques for atypical mycobacteria.
Bronchiectasis may be underdiagnosed in general because it is no longer included in survey data and often goes unreported. The exception is bronchiectasis associated with CF; the latter occurs with a prevalence of 1 in 2500 white births. CF is the largest single cause of chronic lung infections and bronchiectasis in industrialized nations. [66]
Native Americans in Alaska comprise a subgroup with higher-than-expected prevalence, with a 4-fold higher rate of bronchiectasis than the general population. [15] Overall, identifying the true frequency remains a challenge, given the lack of specific symptoms and lack of readily available noninvasive screening tests for population studies.
Race-, sex-, and age-related demographics
No racial predilection exists other than those that may be associated with socioeconomic status.
Evidence suggests that non–CF-related bronchiectasis is more common and more virulent in women, particularly slender white women older than 60 years. In these patients, bronchiectasis is often caused by primary Mycobacterium avium complex (MAC) infection and has been called the Lady Windermere syndrome, named after a character in a novel by Oscar Wilde. [67, 68, 19]
In the preantibiotic era, symptoms usually began in the first decade of life, and this continues to hold true in less-developed countries. Currently, in developed countries, the age of onset has moved into adulthood, except in children with CF. [69]
An epidemiologic study of bronchiectasis-associated hospitalizations in the United States demonstrated that the hospitalization rate for this disorder increased from 1993-2006, especially in persons older than 60 years. [70] No specific single underlying diagnosis has been associated with this apparent increase in the burden of disease in the elderly.
Although limited, epidemiologic studies suggest that persons aged 60-80 years have the highest frequency of bronchiectasis—again likely from the rise in atypical mycobacterial infections. The differences in prevalence between age groups are a direct reflection of the differences in prevalence of the underlying causes of bronchiectasis, lung disease, and/or chronic infections. [71]
Prognosis
In the preantibiotic era, mortality was high, and patients most often died within 5 years after the onset of symptoms. Indeed, a study of 400 patients in 1940 revealed a mortality rate greater than 30%, with most patients dying within 2 years and being younger than 40 years. [72] By comparison, a retrospective study in 1981, after the widespread use of antibiotics, reported a mortality of 13% after diagnosis. [73]
In the late 1990s, researchers in Finland reported no increased mortality in patients with bronchiectasis versus patients with asthma or chronic obstructive pulmonary disease (COPD). Mortality rates for bronchiectasis, asthma, and COPD were 28%, 20%, and 38%, respectively. [74, 75]
Current mortality is difficult to estimate, given the difficulty in identifying prevalence and the lack of definitive studies. Overall, the prognosis for patients with bronchiectasis is good, but it varies with the underlying or predisposing condition. Bronchiectasis associated with CF carries a worse prognosis.
In general, patients do well if they are compliant with all treatment regimens and practice routine preventive medicine strategies. Common complications include recurrent pneumonia requiring hospitalization, empyema, lung abscess, progressive respiratory failure, and cor pulmonale. Additional complications include chronic bronchial infection, and pneumothorax. Life-threatening hemoptysis may occur but is uncommon. Amyloidosis and metastatic abscesses occurred in the preantibiotic era but are rarely observed today.
At present, mortality is more often related to progressive respiratory failure and cor pulmonale than to uncontrolled infection. One study found age older than 65 years and prior use of long-term oxygen therapy to be risk factors for a poor outcome in patients with bronchiectasis who were admitted to an intensive care unit for respiratory failure. [76]
A 2007 study of adults with non-CF bronchiectasis found that higher mortality was associated with advanced age, poor functional status, more severe disease based on radiographic findings, and evidence of hypoxemia or hypercapnia. [77] Preventive care (ie, vaccinations), regular physician visits, and higher body mass index at baseline were associated with reduced mortality.
Patient Education
For patient education information, see the Lung and Airway Center, as well as Chronic Obstructive Pulmonary Disease (COPD).
-
Cylindrical bronchiectasis with signet-ring appearance. Note that the luminal airway diameter is greater than the diameter of the adjacent vessel.
-
Cystic and cylindrical bronchiectasis of the right lower lobe on a posterior-anterior chest radiograph.
-
Varicose bronchiectasis with alternating areas of bronchial dilatation and constriction.
-
This CT scan depicts areas of both cystic bronchiectasis and varicose bronchiectasis.
Tables

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Approach Considerations
- Sputum Analysis
- CBC Count
- Quantitative Immunoglobulin levels
- Quantitative Alpha1-Antitrypsin Levels
- Pilocarpine Iontophoresis (Sweat Test)
- Aspergillus Precipitins and Serum Total IgE levels
- Autoimmune Screening Tests
- Computed Tomography
- Radiography
- Pulmonary Function Tests
- Electron Microscopic Examination
- Bronchography
- Bronchoscopy
- Show All
- Treatment
- Guidelines
- Medication
- Questions & Answers
- Media Gallery
- References







