Practice Essentials
Granulomatosis with polyangiitis (GPA), formerly known as Wegener granulomatosis, is a rare multisystem autoimmune disease of unknown etiology. GPA is one of the antineutrophil cytoplasmic antibody (ANCA)–associated vasculitic disorders. [1] Its hallmark features include necrotizing granulomatous inflammation and pauci-immune vasculitis in small- and medium-sized blood vessels. See the image below.
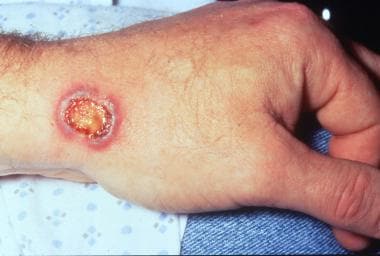 Necrotic, purpuric, and blistering plaque on the wrist in a patient with granulomatosis with polyangiitis.
Necrotic, purpuric, and blistering plaque on the wrist in a patient with granulomatosis with polyangiitis.
Signs and symptoms
GPA has a spectrum of clinical presentations that includes recurrent respiratory infection in adults and upper and lower respiratory tract problems in children. In addition, patients may report the following chronic, nonspecific constitutional complaints:
-
Fevers, night sweats
-
Fatigue, lethargy
-
Loss of appetite
-
Weight loss
Ophthalmic manifestations
-
Conjunctivitis
-
Episcleritis
-
Uveitis
-
Optic nerve vasculitis
-
Retinal artery occlusion
-
Nasolacrimal duct occlusion
-
Proptosis
Ear, nose, and throat (ENT) manifestations
Chronic sinusitis is the most common initial complaint in GPA, occurring in 67% of cases; failure to respond to conventional treatment is suggestive. Other ENT manifestations are as follows:
-
Rhinitis (22%) [2]
-
Epistaxis (11%) [2]
-
Collapse of nasal support, resulting in saddle nose deformity (common)
-
Serous otitis media and hearing loss
-
So-called strawberry gingival hyperplasia
-
Stridor, possibly leading to respiratory compromise, from tracheal or subglottic granulomatous masses
Pulmonary
Pulmonary involvement in GPA can be asymptomatic, insidious in onset, or severe and fulminant. Pulmonary disease may cause any of the following:
-
Pulmonary infiltrates (71%)
-
Cough (34%)
-
Hemoptysis (18%)
-
Chest discomfort (8%) [2]
-
Dyspnea (7%) [2]
-
Diffuse alveolar hemorrhage due to alveolar capillaritis (5%-45%) [3]
-
Atelectasis, with dullness on percussion, decreased breath sounds, and crackles on auscultation
Musculoskeletal manifestations
-
Myalgias
-
Arthralgias, usually polyarticular and symmetrical, affecting small and medium joints
-
Arthritis, typically affecting large joints, but rarely deforming
Renal manifestations
-
Crescentic necrotizing glomerulonephritis characterized by urinary sediment with more than 5 RBCs per HPF or erythrocyte casts
-
Kidney disease is present in 17% of patients at initial diagnosis and is usually asymptomatic [4]
-
Kidney failure occurs in 11% at presentation [2]
Nervous system manifestations
Peripheral nervous system (PNS) involvement may occur in as many as 67% of patients, typically later in the disease course, and includes the following:
-
Mononeuritis multiplex
-
Sensorimotor polyneuropathy
-
Cranial nerve palsies
CNS manifestations include vasculitis of small to medium–sized vessels of the brain or spinal cord and granulomatous masses that involve the orbit, optic nerve, meninges, or brain. [4]
Cutaneous manifestations
-
Cutaneous findings are variable and nonspecific and usually affect the lower extremities
-
Palpable purpura or skin ulcers (45%) [2] ; ulcerations may resemble pyoderma gangrenosum
-
Petechiae, vesicles, pustules, hemorrhagic bullae, livedo reticularis, digital necrosis, subungual splinter hemorrhages, and genital ulcers resembling squamous cell carcinoma have been reported
Additional findings
-
Cardiac: Pericardial rub, myocardial infarction, or sudden death
-
Gastrointestinal: Abdominal pain may be present with splanchnic vasculitis
See Presentation for more detail.
Diagnosis
Routine laboratory tests are nonspecific in GPA. Results may include the following:
-
Abnormal kidney function tests and urinalysis in patients with kidney involvement
-
Rheumatoid factor is positive in a low titer in two thirds of patients
-
CBC: Mild normochromic normocytic anemia is present in 50% of patients; leukocytosis is common, with a neutrophil predominance
-
Elevated inflammatory markers (ESR, CRP)
Antineutrophil cytoplasmic antibody testing
-
Cytoplasmic antineutrophil cytoplasmic antibody (c-ANCA) directed against PR3 is most specific for GPA
-
Some patients with GPA express perinuclear-staining ANCA (p-ANCA) specific for myeloperoxidase (MPO)
-
Combining immunofluorescence and ELISA enhances the sensitivity and specificity of a diagnosis of an ANCA-associated vasculitis (AAV) to 96% and 98.5%, respectively
Chest radiography and CT scanning
-
The most common radiologic findings are single or multiple nodules and masses
-
Nodules are typically diffuse, and approximately 50% are cavitated
-
Diffuse alveolar opacities, atelectasis, and obstructive pneumonia caused by bronchial stenosis may also be seen
-
Findings on CT scans include consolidation, patchy or diffuse ground-glass opacities, or both
-
Additional CT scan findings include stenoses of the larynx or tracheobronchial tree, bronchial wall thickening, bronchiectasis, pleural thickening or effusion, and lymphadenopathy
Other studies
-
Sinus CT scanning: The radiographic test of choice to evaluate sinus disease
-
Pulmonary testing: Spirometry, plethysmography, and diffusing capacity should be performed as soon as possible to identify abnormalities and provide a baseline
-
Bronchoscopy: Helpful in the evaluation of alveolar hemorrhage, infection, airway disease, and endobronchial lesions
-
Biopsy: The diagnosis of GPA is generally confirmed with tissue biopsy from a site of active disease; kidney and lung biopsies are most specific for GPA
See Workup for more detail
Management
Induction of remission in GPA is approached as follows:
-
Cyclophosphamide with glucocorticoids
-
Rituximab with glucocorticoids
-
Low-dose glucocorticoids [5]
-
Methotrexate (oral or subcutaneous) with high-dose glucocorticoids, in non–organ-threatening or non–life-threatening GPA [6]
Maintenance of remission
-
Once induction of remission has occurred, treatment for maintenance of remission should be continued for at least 18 months, often longer
-
Azathioprine (2 mg/kg/day) is safer than, and as effective as, cyclophosphamide in maintaining remission [8]
-
Methotrexate (20-25 mg weekly, oral or subcutaneous) has been used for the maintenance of remission if the serum creatinine level is less than 1.5 mg/dL
-
Leflunomide (20-30 mg/day) is as effective as methotrexate, but it is associated with more adverse effects [9]
See Treatment and Medication for more detail.
Background
In 1897, Peter McBride likely gave the first written description of a patient with granulomatosis with polyangiitis (GPA). In 1931, Klinger described a 70-year-old physician with constitutional symptoms, joint symptoms, proptosis, widespread upper respiratory tract inflammation leading to saddle nose deformity, glomerulonephritis, and pulmonary lesions.
In 1936, Dr. Frederich Wegener reported three patients with similar clinical features and published his findings on their distinct clinical and histopathologic findings, leading to the eponymous designation of the disease.
In 1954, Goodman and Churg provided the definitive description of GPA upon their identification of a triad of pathological features that characterize the disease, including (1) systemic necrotizing angiitis, (2) necrotizing granulomatous inflammation of the respiratory tract, and (3) necrotizing glomerulonephritis. (See Etiology, Presentation, and Workup.)
The American College of Rheumatology, European League Against Rheumatism, and American Society of Nephrology have recommended a gradual shift from honorific eponyms to disease-descriptive or etiology-based nomenclature. In the case of GPA, the change was triggered by evidence that Wegener was a member of the Nazi party before and during World War II. The recommended alternative name was already being used in the medical literature. [10]
Before the institution of effective therapy, the mean survival of adults with untreated GPA was only 5 months, with 82% of patients dying within the first year and 90% of patients dying within the second year. Despite improvement with the use of corticosteroids, the mean survival time was increased only to 12.5 months.
With the advent of cytotoxic therapy for GPA, patient survival markedly improved. In 1983, Fauci et al reported a 93% complete remission rate in 85 patients (mean age 43.6 y, range 14-75 y) treated with prednisone and oral cyclophosphamide. [2]
2022 criteria for the classification of granulomatosis with polyangiitis
These criteria from the American College of Rheumatology and the European Alliance of Associations for Rheumatology are used for enrolling patients in studies and should not be considered as diagnostic criteria. [11] The classification criteria should be used when a diagnosis of small- or medium-vessel vasculitis has been made, and alternative disorders mimicking vasculitis have been excluded. The criteria are divided into clinical criteria and laboratory, imaging, and biopsy criteria. Each criterion is assigned a score.
Clinical criteria and scores are as follows:
-
Nasal involvement – Bloody discharge, ulcers, crusting, congestion, blockage, or septal defect/perforation: +3
-
Cartilaginous involvement – Inflammation of ear or nose cartilage, hoarse voice or stridor, endobronchial involvement, or saddle nose deformity: +2
-
Conductive or sensorineural hearing loss: +1
Laboratory, imaging, and biopsy criteria and scores are as follows:
-
Positive test for cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) or antiproteinase 3 (anti-PR3) antibodies: +5
-
Pulmonary nodules, mass, or cavitation on chest imaging: +2
-
Granuloma, extravascular granulomatous inflammation, or giant cells on biopsy: +2
-
Inflammation, consolidation, or effusion of the nasal/paranasal sinuses, or mastoiditis on imaging: +1
-
Pauci-immune glomerulonephritis on biopsy: +1
-
Positive test for perinuclear antineutrophil cytoplasmic antibodies (p-ANCA) or antimyeloperoxidase (anti-MPO) antibodies: -1
-
Blood eosinophil count ≥ 1 × 10 9/L: -4
For the purposes of classification, a patient with a total score of 5 or more is said to have GPA. These criteria have a sensitivity of 92.5% and a specificity of 93.8%. [12]
Limited versus severe granulomatosis with polyangiitis
GPA is one of the ANCA-associated vasculitides (AAVs) and has a predilection for the upper and lower respiratory tracts and the kidneys. It has a spectrum of clinical presentations and may be divided broadly into limited or severe disease.
Individuals with limited GPA present with clinical findings largely isolated to the upper and lower respiratory tracts and are generally not considered to have organ- or life-threatening disease. Persons with severe disease present with significant multisystem manifestations that may involve the lungs, kidneys, and other organs, in addition to the respiratory tract. Severe disease can also be described as generalized disease.
Consensus does not exist as to whether limited GPA represents early severe disease or an altogether separate clinical entity. The terminology, limited versus severe, can sometimes be problematic because pulmonary and/or kidney involvement may be absent at the onset of symptoms.
Longitudinal follow-up of the National Institutes of Health GPA cohort (158 patients who were observed for 6 mo to 24 y) demonstrated that 18% of patients initially had kidney disease and that 77% had glomerulonephritis upon later analysis. [4] Thus, patients initially diagnosed with limited GPA may subsequently develop generalized disease with kidney involvement.
Analysis of another GPA cohort, that of the Wegener Granulomatosis Etanercept Trial (WGET), suggested that limited disease may be a qualitatively different clinical entity. In the WGET cohort, patients classified as having limited disease had more severe upper respiratory tract damage, were more likely to have flares after remission, and tended to have identical manifestations when they relapsed. [13]
Below is an outline of the differences between limited and severe GPA based on the WGET trial manual of operations.
Limited granulomatosis with polyangiitis
This designation is reserved for cases that fulfill the 1990 American College of Rheumatology criteria for the classification of GPA, [12] but lack disease features that pose immediate threats to either a critical individual organ or to the patient's life. [13] Specifically, this means the following:
-
The patient has no RBC casts in the urine.
-
If hematuria is present (but no RBC casts), the serum creatinine level is 1.4 mg/dL or less, and the creatinine level has not risen more than 25% above the patient’s baseline level.
-
Pulmonary involvement must be circumscribed, such that the room air partial pressure of oxygen (PO2) level is greater than 70 mm Hg or the room air O2 saturation by pulse oximetry is greater than 92%; pulmonary hemorrhage may be treated as limited disease provided that there is no evidence of progression of process (in the absence of data on progression, pulmonary hemorrhage may be treated as severe disease at the discretion of the physician)
-
No disease exist within any other critical organ (eg, the gastrointestinal [GI] tract, eyes, central nervous system [CNS]) that, without the immediate institution of maximal therapy (ie, pulse methylprednisolone or daily oral cyclophosphamide), threatens the function of that organ and/or the patient’s life.
Severe granulomatosis with polyangiitis
Any patient with GPA whose disease is not classifiable as limited has severe disease, by definition. [13]
Etiology
The pathologic hallmarks of GPA are vasculitis of the small- to medium-sized vessels, "geographic" necrosis, and granulomatous inflammation, particularly in the airways. The initial pathologic lesion is that of the granuloma believed to be caused by cellular immune processes.
Environmental exposures, including respiratory tract infections, have been implicated as inciting factors for granuloma formation. A better understanding of the progression from granuloma to vasculitis may shed light on the possible etiology and pathogenesis of GPA. It is probable that a complex interaction exists between the environment and host factors, many of which are genetically determined. Cellular immune processes are also involved in tissue injury owing to the inflammatory cascade.
ANCAs
The discovery of ANCAs within neutrophils in the majority of patients with GPA suggested the role of humoral autoimmunity. GPA is usually associated with the presence of diffuse staining cytoplasmic ANCA (C-ANCA) directed against serine proteinase 3 antigen (PR3-ANCA), the so-called Wegener autoantigen.
The other AAVs include microscopic polyangiitis, renal-limited vasculitis, and Churg-Strauss syndrome (allergic granulomatous angiitis), which are more commonly associated with perinuclear-staining ANCA (P-ANCA) directed against myeloperoxidase (MPO-ANCA).
A pathogenic role for PR3-ANCAs in GPA has been proposed, because PR3-ANCA is strongly associated with the disease; over 90% of GPA patients have been reported to have ANCA positivity during active disease. [14] Longitudinal observations have indicated that relapse is sometimes heralded by a rise in PR3-ANCA titers, although other studies could not confirm these results. [15, 16, 17]
Schlieben et al reported that a newborn developed a pulmonary-renal syndrome associated with transplacental passage of MPO-ANCA immunoglobulin G (IgG) from a mother with ANCA disease who developed a clinical and serologic flare of disease during pregnancy. [18]
Another argument for the pathogenic role of PR3-ANCA comes from observations that ANCA persistence after induction of remission in patients with GPA is associated with relapse. [19] Additionally, efficacy of treatment with rituximab, a B-cell depleting monoclonal antibody and, thus, an inhibitor of antibody production, supports a pathogenic role of ANCA in patients with AAV. [20, 21]
Evidence also comes from in vitro studies. The in vitro effects of PR3-ANCA described to date include activation of primed neutrophils, leading to production of reactive oxygen species, and release of lytic enzymes such as elastase and PR3, which act to promote tissue injury. [22, 23] In vitro data also demonstrate the role of complement in AAV and show that ANCAs are involved in neutrophil-endothelial cell activation. Both of these processes likely help to target the endothelium, resulting in necrotizing vasculitis. [24]
In vivo experimental studies have demonstrated a pathogenic role for MPO-ANCA in mice and rat models. MPO-ANCA induces a pauci-immune necrotizing glomerulonephritis and hemorrhagic capillaritis in these animal models. [25] Neutrophils, as well as the complement system, are necessary for lesion development. [24] Despite this in vivo evidence for a pathogenic role of MPO-ANCA in AAV-like syndromes in animal models, no definitive in vivo evidence has yet been found for PR3-ANCA. Further research is necessary to further elaborate its role in GPA.
Genetics
Typically, most autoimmune diseases are attributed to a genetic predisposition combined with exposure to an inciting factor. Genotypic associations in GPA include the following [22, 26, 27, 28, 29, 30, 31] :
-
Carrying a defective allele for alpha-1 antitrypsin
-
Possessing certain polymorphisms of CTLA-4 (cytotoxic T-lymphocyte antigen 4), which is involved in T-cell activation
-
Possessing the PTPN22*620W allele, which is typically associated with a positive ANCA status and is also associated with T-cell activation
-
Carrying the DPB1*0401 allele, which is also associated with chronic beryllium disease, a granulomatous disease
-
Exhibiting certain forms of the Fcγ receptor IIIb on the surface of neutrophils and monocytes/macrophages.
Microbes
The role of microbes in the pathogenesis of AAV has also been explored, although the mechanism has not been fully explained. The first evidence for this was discovered by Stegeman et al, who noted that nasal carriage of Staphylococcus aureus is associated with relapses of GPA (relative risk, 9.0) and prophylactic treatment with trimethoprim-sulfamethoxazole (TMP-SMZ) can reduce the likelihood of relapse by 60%. [19]
Since then, there has been the discovery that complementary PR3, which shows homology with certain S aureus–derived peptides, may induce antibodies to PR3. [32] Additionally, at least in rat models, infection with gram-negative bacteria may lead to the development of AAV in susceptible individuals. [33]
Other factors
Living in northern latitudes, farming, drug and environmental allergies, and exposures to solvents or silica have all been linked to the development of GPA. [28, 34] Reports vary as to whether disease onset has a seasonal peak.
Epidemiology
GPA is a rare disease with an as yet undetermined incidence. The prevalence of GPA in the United States is estimated to be 3 cases per 100,000 people.
The incidence and prevalence of GPA in the United Kingdom is estimated at 10.2 cases and 250 cases per million population, respectively.
Race-, sex-, and age-related demographics
GPA is more common in individuals of northern European descent (approximately 90%); it occurs less commonly in blacks.
In European populations, GPA is slightly more common in men, with a male-to-female ratio of 1.5:1. [35] Women are more likely to have limited disease. [13]
The onset of GPA may occur at any age, although patients typically present at age 35-55 years. GPA is rare in childhood. [36] In a systematic review and meta-analysis of childhood-onset GPA, the age range was 4-18 years. The majority of patients were adolescent girls. [37]
Prognosis
The remission rate in GPA ranges from 30-93%, depending on the definition of remission and the remission induction therapy used. [38] With aggressive therapy for active disease, more than 50% of patients with GPA recover renal function and are able to become dialysis independent. [39, 40]
Unfortunately, relapse is common in GPA. Typically, up to half of patients with GPA experience relapse within 5 years. [41] The rate (18-40% at 24 mo) and time to first relapse (15-29 mo) varies. [38] Factors associated with relapse include treatment (< 10 g of cyclophosphamide in the first 6 mo, maintaining a high dose of prednisone [>20 mg/day] for < 2.75 mo, and goal of 0 dose of glucocorticoids), ANCA status at diagnosis, and target organ involvement (lung involvement, cardiac involvement, renal involvement, chronic nasal carriage of S aureus). [38, 42]
Rising PR3-ANCA (C-ANCA) titers may correlate with disease activity in approximately two-thirds of patients. However, the relationship is unreliable; thus, negative PR3-ANCA results do not necessarily exclude the possibility of relapse. [41] As significant adverse effects are associated with immunosuppressive therapy, especially cyclophosphamide, ANCA persistence or reappearance should be used as a warning signal rather than an indication to escalate therapy.
Poorer survival is associated with older age, target organ involvement, and target organ damage. Renal involvement has been consistently shown to confer a poorer prognosis. An absence of kidney involvement is associated with a 100% 5-year survival rate, compared with approximately 70% in individuals with kidney disease. [43] An increased risk of cardiovascular events is also noted. Overall, the 10-year survival rate ranges from 75-88%. [43] Most morbidity in GPA is currently treatment related.
Complications
In a longitudinal cohort consisting of 158 patients with GPA, from the National Institutes of Health (NIH), 86% of patients experienced permanent damage from their disease. [44] Permanent damage includes the following:
-
End-stage kidney disease
-
Chronic pulmonary dysfunction
-
Hearing loss
-
Destructive sinus disease
-
Saddle nose deformities
-
Perforation of the nasal septum
-
Proptosis
-
Blindness
Respiratory problems may result from upper-airway obstruction (eg, subglottic stenosis) or pulmonary involvement (eg, pleural effusion, dyspnea, diffuse alveolar hemorrhage [DAH]).
Many patients in the NIH cohort (42%) also experienced permanent treatment-associated morbidity, including hemorrhagic cystitis, osteoporotic fractures, urothelial (bladder) cancer, myelodysplasia, and avascular necrosis. [44] Urotoxic adverse events associated with cyclophosphamide use are related to cumulative dose and oral administration. Cyclophosphamide treatment of systemic vasculitis increases the risk of urothelial cancer 5-fold over that of the general population. [45]
Furthermore, the development of other cancers associated with immunosuppression in patients with AAV is a concern, as it is for patients with other inflammatory rheumatologic and nonrheumatologic diseases and for patients who have undergone organ transplantation. Increased rates of leukemia, lymphoma, and nonmelanoma skin cancers have been reported in a number of studies of treated patients with AAV. The observed overall incidence of cancers in this population is 1.6-2.4 times higher than in the general population. [46] Clinicians caring for these patients should keep this increased risk in mind and refer and/or screen appropriately.
Additionally, an increased rate of cardiovascular events is noted in patients with AAV. A European study that reviewed outcomes during long-term follow-up of patients with GPA and microscopic polyangiitis determined that within 5 years of diagnosis, 14% of patients experienced at least 1 cardiovascular event. This was 3.7 times higher than was expected in the background population. This study determined that older age, diastolic hypertension, negative PR3-ANCA status, and positive MPO-ANCA status are independent determinants of cardiovascular outcomes in patients without prior cardiovascular disease. [47]
A study of a Canadian population-based database with newly diagnosed GPA reported a hazard ratio (HR) of 1.86 for myocardial infarction (MI) and 1.50 for ischemic stroke. The HR for cardiovascular disease (composite outcome of MI or stroke) was highest during the first year after GPA diagnosis (HR 2.88). [48]
Mortality
Severe, untreated GPA is associated with a very high (>90%) mortality rate. Historically, patients with untreated GPA had a mean survival of 5 months from diagnosis; the mortality rate was 82% at 1 year. The introduction of corticosteroids prolonged the median survival by only 7.5 months.
With the advent of cytotoxic therapy, patient survival in GPA markedly improved. According to a meta-analysis, with current treatments, the 5-year survival rate ranges from 74-79%. [43] The 1-year mortality rate is still high, around 11% (range, 2.2-25%), depending on disease severity and the intensity of treatment. [49] In a study of a US inpatient database, the annual hospitalization rate for patients with a principal diagnosis of GPA increased by 24% from 1993 to 2011, from 5.1 to 6.3 per 1,000,000 population; however, in-hospital deaths durig that period declined 73%, from 9.1% to 2.5%. [50]
The most common causes of death in GPA are as follows:
-
Infection
-
Respiratory and kidney failure
-
Malignancy
-
Cardiovascular events
Patient Education
Patients with GPA and their families must be educated on the serious nature of this disease. Potential risks and adverse effects of immunosuppressive medications should be detailed. For patient education information, see Granulomatosis with Polyangiitis Symptoms, Treatment, and Life Expectancy. Patient education information is also available from the American College of Rheumatology and the Vasculitis Foundation.
-
Saddle nose deformity in a 26-year-old man with granulomatosis with polyangiitis. Image courtesy of P. Papadopoulos, MD.
-
Necrotic, purpuric, and blistering plaque on the wrist in a patient with granulomatosis with polyangiitis.
-
Several necrotic, purpuric, and blistering papules and plaques on the hands in a patient with granulomatosis with polyangiitis.
-
Granulomatosis with polyangiitis (formerly known as Wegener granulomatosis). Large ulceration of the pharynx covered with a dense necrotic membrane.
-
Extensive thickening of the maxillary sinuses in a patient with granulomatosis with polyangiitis. The patient also had intermittent epistaxis.
-
Granulomatosis with polyangiitis. Bilateral nodules observed on a plain chest radiograph in a patient with hemoptysis and hematuria. Image courtesy of G. Eschun, MD.
-
This 42-year-old man presented with hemoptysis, weight loss, and night sweats. He was diagnosed with the limited form of granulomatosis with polyangiitis.
-
Bilateral cavitating nodules in a patient with granulomatosis with polyangiitis.
-
Granulomatosis with polyangiitis. This patient presented with massive hemoptysis. No nodules are identified on the chest radiograph, although a subsequent CT scan showed several noncavitating nodules.
-
Diffuse alveolar hemorrhage in a 21-year-old man with granulomatosis with polyangiitis. Image courtesy of the US Government.
-
Diffuse alveolar hemorrhage in a 21-year-old man with granulomatosis with polyangiitis. Image courtesy of the US Government.
-
Shown is a chest radiograph of an 11-year-old girl who presented with an upper respiratory tract infection, myalgias, and arthralgias for 1 month, followed by an abrupt presentation with pallor, hemoptysis, and hypertension. Her bilateral fluffy infiltrates are suggestive of a pulmonary hemorrhage. She had an antineutrophil cytoplasmic autoantibody (ANCA)–positive pauci-immune necrotizing and crescentic glomerulonephritis associated with her pulmonary hemorrhage. Supportive therapy consisted of mechanical ventilation and hemodialysis along with immunosuppressive therapy. Her anti–glomerular basement membrane antibody test result was negative. Nearly 2 years later, she had a serum creatinine of 0.7mg/dL and no residual pulmonary disease.
-
An 11-year-old girl presented with an upper respiratory tract infection, myalgias, and arthralgias for 1 month followed by an abrupt presentation with pallor, hemoptysis, and hypertension. She had an antineutrophil cytoplasmic autoantibody (ANCA)–positive pauci-immune necrotizing and crescentic glomerulonephritis associated with her pulmonary hemorrhage. A follow-up chest radiograph obtained several days later shows a complete resolution of her pulmonary infiltrates. This rapid resolution is more consistent with hemorrhage than with pneumonia.
-
A renal biopsy specimen from a 13-year-old girl with antineutrophil cytoplasmic antibody (C-ANCA)–positive pulmonary renal syndrome. Seven weeks after presenting with sinusitis, she presented with an acute abdomen, pulmonary hemorrhage, and acute renal failure (creatinine 4.9mg/dL). This biopsy specimen shows a necrotizing and crescentic glomerulonephritis (Silver stain).
-
Lung biopsy specimen from a patient with granulomatosis with polyangiitis showing evidence of vasculitis and inflammation (high-power view). Image courtesy of Z. Xu, MD.
-
Lung biopsy specimen from a patient with granulomatosis with polyangiitis showing evidence of vasculitis and inflammation (high-power view). Image courtesy of Z. Xu, MD.
-
Focal glomerulonephritis with crescent formation on renal biopsy specimen, characteristic of granulomatosis with polyangiitis.
-
C-ANCA immunofluorescence pattern. Staining for antineutrophil cytoplasmic antibody by indirect immunofluorescence shows heavy cytoplasmic staining, whereas nuclei are nonreactive. Image courtesy of K. Orr, MD.
-
P-ANCA immunofluorescence pattern. Perinuclear antineutrophil cytoplasmic antibody staining pattern by indirect immunofluorescence shows perinuclear staining, whereas cytoplasm is nonreactive. Image courtesy of K. Orr, MD.
-
Goodpasture syndrome: Linear deposition of immunoglobulin G and C3 are observed on a renal biopsy specimen from a patient with Goodpasture syndrome. Immunofluorescence staining. Image courtesy of K. Orr, MD.






