Background
Ventricular fibrillation (VF) is a life-threatening cardiac arrhythmia in which the coordinated contraction of the ventricular myocardium is replaced by high-frequency, disorganized excitation, resulting in [the effective] failure of the heart to pump blood. VF is the most commonly identified arrhythmia in cardiac arrest patients. In the prehospital setting, 65%-85% of patients in cardiac arrest have VF identified as the initial rhythm by emergency services personnel. [1, 2, 3] (See Presentation and Workup.)
VF usually ends in death within minutes unless prompt corrective measures are instituted. The rate of survival in out-of-hospital cardiac arrest has increased with the expansion of community-based emergency rescue systems, widespread use of automatic external defibrillators (AEDs), and increasing numbers of laypersons trained in bystander cardiopulmonary resuscitation (CPR), but it nonetheless remains low. (See Prognosis, Treatment and Medication.)
In hospital settings, VF is treated using Advanced Cardiac Life Support (ACLS) protocols. Long-term management may be accomplished with medical therapy or placement of an implantable cardioverter-defibrillator (ICD). Surgical correction of underlying disorders (eg, percutaneous coronary intervention, coronary artery bypass surgery) may also be indicated. (See Treatment and Medication.)
For related topics, see Ventricular Fibrillation in Emergency Medicine, Sudden Cardiac Death, Hypertrophic Cardiomyopathy, and Pediatric Ventricular Fibrillation.
Patient education
For patient education information, see the Heart Health Center, Cholesterol Center, and Healthy Living Center, as well as Atrial Fibrillation (A Fib), Chest Pain, Arrhythmias (Heart Rhythm Disorders), Heart Disease, Heart Attack, Cardiopulmonary Resuscitation (CPR), and Tetralogy of Fallot.
Pathophysiology
Ventricular fibrillation (VF) occurs in a variety of clinical situations but is most often associated with coronary artery disease (CAD). VF can result from acute myocardial infarction (MI) or ischemia, or from myocardial scarring from an old infarct. [4] Ventricular tachycardia (VT) may degenerate into VF. Intracellular calcium accumulation, the action of free radicals, metabolic alterations, and autonomic modulation are important influences on the development of VF during ischemia.
Initiation of VF can occur in several ways. For example, if the myocardium is stimulated by a ventricular premature complex (VPC) during the ascending limb of the T wave, [5] the impulse can propagate erratically through the variably refractory myocardial cells and establish reentrant patterns that result in chaotic ventricular depolarization. Consequently, coordinated myocardial contraction becomes disrupted.
The reentrant patterns break up into multiple smaller wavelets and the level of disorganization increases, with reentrant circuits producing high-frequency activation of cardiac muscle fibers. As the heart loses its ability to pump blood, myocardial ischemia worsens and a self-perpetuating vicious cycle ensues, leading to death if not corrected.
On the electrocardiogram (ECG), VF manifests as a chaotically irregular pattern. This pattern is coarse initially but becomes finer as ventricular disorganization increases. As the ECG waveform flattens, the likelihood of successful defibrillation decreases. [6]
Etiology
Coronary artery disease (CAD) is the single most common etiologic factor predisposing patients to ventricular fibrillation (VF). In survivors of cardiac arrest, CAD with over 75% stenosis is observed in 40%-86% of patients, depending on the age and sex of the population studied. In postmortem studies of people who have died from VF, extensive atherosclerosis is the most common pathologic finding.
In an autopsy study of 169 cases of coronary death, approximately 61% of patients had died suddenly of presumed VF, and another 15% of cases showed more than 75% stenosis in three or four vessels as well as similar severe lesions in at least two vessels. [7] No single coronary artery lesion is associated with an increased risk for VF.
Nevertheless, only approximately 20% of VF-related autopsies have shown evidence of a recent myocardial infarction (MI). A greater proportion of autopsies (40%-70%) show evidence of a healed MI. Many of these hearts also reveal evidence of plaque fissuring, hemorrhage, and thrombosis. [8]
In young people, causes of autopsy-positive sudden cardiac death (SCD) include hypertrophic cardiomyopathy (HCM) and arrhythmogenic right ventricular dysplasia (ARVD), whereas inherited arrhythmogenic etiologies cause autopsy-negative SCD, including long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), Wolff- Parkinson-White (WPW) syndrome, idiopathic VF, and Brugada syndrome. [9]
The Coronary Artery Surgery Study (CASS) showed that surgically improving or restoring blood flow to the ischemic myocardium decreased the risk of VF, especially in patients with three-vessel disease and heart failure, compared with medical treatment. [10] This finding suggests that transient acute ischemia is one of the major triggering events for sudden arrhythmic death.
The efficacy of beta-blocking agents in reducing sudden death mortality rates, especially when administered to patients suffering from MI with VF, ventricular tachycardia (VT), and high-frequency premature ventricular contractions (PVCs), is thought to be partially caused by the ability of beta blockers to decrease ischemia. Beta blockers also increase the VF threshold in ischemic animals and reduce the rate of ventricular ectopy in patients with MI.
Reperfusion of ischemic myocardium with thrombolysis or angioplasty can induce transient electrical instability by several different mechanisms. One of these, coronary artery spasm, exposes the myocardium to both ischemia and reperfusion insults. Possible mechanisms of coronary vasospasm include autonomic nervous system factors, especially alpha-adrenergic activity; vagal activity; vessel susceptibility; and humoral factors, particularly those associated with platelet activation and aggregation.
Nonatherosclerotic coronary artery abnormalities are also associated with an increased incidence of sudden death. Such abnormalities include congenital lesions, embolism, arteritis, and mechanical abnormalities, such as coronary artery aneurysms.
When documentation of the antecedent rhythm is available, it often shows that rapid VT precedes VF. In patients with chronic ischemic heart disease, monomorphic VT arising from a reentrant focus is the most common precursor to VF. Other factors associated with an increased risk of VF include frequent PVCs, particularly complex forms (such as multiform PVCs) and ones with short coupling intervals (R-on-T phenomenon). [11]
Even though many individuals have anatomic and functional cardiac substrates that predispose them to ventricular arrhythmias, only a small percentage develop VF. The interplay among regional ischemia, left ventricular (LV) dysfunction, and transient inciting events (eg, worsened ischemia, acidosis, hypoxemia, wall tension, drugs, metabolic disturbances) has been proposed to be the precipitator of VF.
An estimated 3%-9% of cases of VT and VF occur in the absence of myocardial ischemia. Up to 1% of patients with out-of-hospital cardiac arrest have idiopathic VF with no discernable structural heart disease. [12] Up to 15% of patients younger than 40 years who experience VF have no underlying structural heart disease. Belhassen and Viskin noted that 4 of 11 patients in their study with a history of VF and no structural heart disease had histologic abnormalities on endomyocardial biopsy. [13]
Acute and chronic ischemic heart disease
Cardiac arrest attributable to ventricular arrhythmias may occur with acute ischemia or in the absence of an acute disturbance of coronary flow, due to scarring from a previous MI. An infarct scar can serve as the focus for reentrant ventricular tachyarrhythmias, which may occur shortly after the infarct or years later. Many studies support the relationship of symptomatic and asymptomatic ischemia as markers of myocardium at risk for arrhythmias. [14, 15]
Patients resuscitated from out-of-hospital cardiac arrest are at an increased risk for recurrent cardiac arrest and express an increased incidence of silent ST-segment depressions. [16] In animal models, experimentally induced myocardial ischemia has shown a strong relationship with the development of VF.
Nonischemic cardiomyopathies
Patients with nonischemic cardiomyopathies are the second largest group of patients who experience VF, accounting for approximately 10% of VF cases. Nonischemic myopathies, for the purposes of this article, can be divided into dilated, hypertrophic, and other, rarer, forms. These cardiomyopathies can predispose to both VT and VF. The VT can degenerate into VF, persist as a rhythm that is stable enough to allow detection and directed therapy, or terminate spontaneously, with or without associated symptoms.
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is recognized more frequently, with a reported annual incidence of approximately 7.5 cases per 100,000 people. The prognosis after a VF event is very poor for these patients, with a 1-year mortality rate of 10%-50%, depending on the New York Heart Association functional class; VF causes approximately 30%-50% of these deaths.
DCM has varied etiologies, including idiopathic, viral, autoimmune, genetic, or environmental (eg, alcohol abuse). The predominant mechanism of sudden death in patients with DCM appears to be ventricular tachyarrhythmias, although bradyarrhythmias and electromechanical dissociation have also been observed, especially in patients with advanced LV dysfunction. [17] Extensive subendocardial fibrosis leading to ventricular dilatation and subsequent generation of reentrant tachyarrhythmias is a proposed substrate for VF.
Multiple factors contribute to an increased risk for VF in this population. The most important hemodynamic predictor is an increase in LV end-diastolic pressure and the resulting increased wall tension. Other important factors are an increased sympathetic tone, neurohumoral activation, and electrolyte abnormalities. Many drugs used in the treatment of heart failure, such as antiarrhythmics, inotropic agents, and diuretics, have proarrhythmic properties, which may provoke arrhythmias in some patients.
The genetic causes of DCM are myriad, with many genes, including those that encode actin, myosin, and troponin proteins, being implicated in its causation. Most familial DCMs are inherited in an autosomal dominant fashion, with mutations typically occurring in the proteins found in the cardiac sarcomere.
Interestingly, genes such as PSEN1 and PSEN2, which are responsible for early onset Alzheimer disease, have also been implicated in DCM. X-linked inheritance of DCM has been described in patients with mutations in the DMD (Duchenne muscular dystrophy) gene and the TAZ (Barth Syndrome) gene. Autosomal recessive inheritance has been described in mutations of the TNNI3 gene, which encodes the troponin I muscle protein.
Hypertrophic cardiomyopathy
HCM is usually an autosomal dominant, incompletely penetrant genetic disorder resulting from a mutation in one of the many (>45) genes that encode proteins of the cardiac muscle sarcomere. [18] Among the described genetic abnormalities are mutations in the genes encoding the beta-myosin heavy chains (MYH7), cardiac troponin T (TNNT2), myosin-binding protein C (MYBPC3), and cardiac troponin I.
Mutations in these four genes account for approximately 90% of HCM. The incidence of VF in this population is 2%-4% per year in adults and 4%-6% per year in children and adolescents. HCM is the most common cause of VF before age 30 years.
The mechanism of VF in HCM is not entirely understood. Ventricular arrhythmias in HCM are probably a result of a substrate of electrical instability and disorganized electrophysiologic (EP) transmission due to abnormal LV myocardial architecture. Intramural CAD leading to episodic myocardial ischemia and the resulting necrosis and fibrosis has also been implicated as a potential substrate for VT/VF. [19]
The vast majority of young people who die of HCM are previously asymptomatic. Many experience VT/VF while at rest or with mild exertional activity; however, in a significant portion of these patients, the VF event occurs after vigorous exertion. The postexertional drop in blood pressure and shunting of blood to extracardiac tissues is postulated to worsen the outflow tract gradient and may therefore induce cardiac ischemia and malignant arrhythmias.
This downward cycle does not revert spontaneously, and it responds poorly to resuscitative efforts. HCM is the single greatest cause of VF in athletes and is therefore the major entity to screen for during the physical examination of an athlete. [18, 20]
Arrhythmogenic right ventricular cardiomyopathy/dysplasia
Arrhythmogenic right ventricular (RV) cardiomyopathy/dysplasia (ARVC/D) is characterized by replacement of the RV wall with fibrofatty tissue. Involvement of the interventricular septum is unusual, and involvement of the left ventricle is associated with poorer outcomes. [21, 22]
The genetics of ARVC/D are extremely heterogeneous. At least 10 genes (TGFB3, RYR2, DSP, PKP2, DSG2, TMEM43, JUP, TTN, DES, DSC2) [23] and four additional loci (14q12-q22, 2q32.1-32.3, 10p14-p12, and 10q22) have been implicated in the pathogenesis of this disorder, which is inherited in an autosomal dominant fashion with incomplete penetrance. Eight of these genes are thought to be responsible for approximately 42.5% of the total cases of ARVC/D. [24]
The majority of the genetic mutations relate to desmosomal abnormalities. The desmosomes are proteins that are instrumental in cell-to-cell binding of myocytes.
Patients with ARVC/D, which is more prevalent in men than in women, may present with signs and symptoms of RV hypertrophy and dilatation, often with sustained monomorphic VT with left bundle-branch block morphology, with an axis usually between negative 90°-100°. Less commonly, patients present with polymorphic VT. Atrial arrhythmias may be present in up to 25% of patients. The annual incidence rate of VF in patients with ARVC/D is approximately 2%.
Syncope and sudden death in ARVC/D are often associated with exercise. In many patients, sudden death—the incidence of which is highest in persons aged 30-50 years—is the first manifestation of the disease. Symptoms are rare in preadolescent children.
The most common electrocardiographic (ECG) abnormality in ARVC/D is T-wave inversion in leads V1 -V3. Epsilon waves are seen in V1 or V2 as sharp spikes in the ST segment. In V1, a delayed-onset S wave (from nadir to baseline >60 msec) is a specific sign of ARVC/D (see the image below). In addition, PVCs with a left bundle-branch block pattern are common. Signal-averaged ECGs are abnormal in the majority of patients.
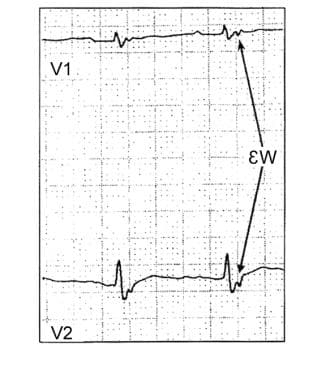 This image shows an epsilon wave on the electrocardiogram of a patient with arrhythmogenic right ventricular dysplasia (ARVD).
This image shows an epsilon wave on the electrocardiogram of a patient with arrhythmogenic right ventricular dysplasia (ARVD).
ARVC/D differs from Uhl anomaly, a condition in which the RV wall is extremely thin because of apposition of the endocardial and epicardial layers. Uhl anomaly usually manifests in the pediatric population, whereas ARVC usually manifests in adults.
The diagnosis can be confirmed with echocardiography or, preferably, with cardiac magnetic resonance imaging (CMRI) studies. Voltage mapping of the RV can be performed using a three-dimensional (3-D) mapping system and can show the presence of low-voltage areas (surrogate for scar or fatty infiltration) in the so-called triangle of dysplasia.
Because there is no gold standard for the diagnosis of ARVC/D, clinicians resort to the use of a variety of clinical abnormalities known as major and minor criteria. The diagnosis of ARVC/D is thought to be definite if the patient has any of the following [25, 26] :
-
At least two (2) major criteria
-
One (1) major and two (2) minor criteria
-
Four (4) minor criteria
Valvular lesions
Aortic stenosis
Before the advent of surgical therapy for valvular heart disease, SCD was fairly common in patients with progressive aortic stenosis. Chizner et al followed 42 patients with isolated, unoperated aortic stenosis for over 5 years and found that symptomatic patients had a high mortality rate and that 56% of deaths occurred suddenly (within hours of the development of new symptoms). However, there were no deaths in asymptomatic patients. [27]
The mechanism of SCD in patients with uncorrected aortic stenosis is unclear, although malignant tachyarrhythmias and bradyarrhythmias have been documented. VF accounts for up to 20% of deaths after aortic valve replacement and remains the second most common cause of postoperative death in this population. The incidence of VF after aortic valve surgery is highest in the first 3 weeks after the procedure and then plateaus at 6-month follow-up.
Other
Patients with chronic aortic insufficiency usually present with signs of chronic heart failure due to progressive LV dilatation and remodeling. As part of this process, reentrant or automatic ventricular foci may develop and cause symptomatic ventricular arrhythmias. After valve replacement, LV wall tension can be expected to lessen, and the risk of arrhythmia can be expected to decrease.
Mitral stenosis has become uncommon in the United States because of widespread use of antibiotics in primary streptococcal infections. SCD due to mitral stenosis is very rare.
The incidence of VF is low in patients with mitral valve prolapse (MVP). In clinically significant MVP, the risk of VF seems to rise along with the total mortality rate. The incidence of sudden death appears to vary with the presence of symptoms and the severity of mitral regurgitation. MVP has a 5%-7% occurrence rate in the general population. [28]
Congenital structural heart disease
The causes of SCD are much more diverse in children than in adults. In reviewing 13 studies, involving 61 children and adolescents with VF, Driscoll and Edwards found that 50% of cases were due to HCM and that 25% resulted from anomalous origin of the left coronary artery. [29] The remaining cases were due to aortic stenosis, cystic medial necrosis, and sinus node artery obstruction.
Disease entities associated with VF in patients with known, previously recognized (including repaired) congenital heart disease include the following:
-
Transposition of the great arteries
-
Physiologic single ventricle
-
Congenital heart block
The predominant mechanism is ventricular arrhythmia. In patients with tetralogy of Fallot, up to 10% have VT after surgical correction of the anomaly, and the incidence of sudden death is 2%-3%. In the Fontan procedure to correct a physiologic single ventricle, even atrial arrhythmias can cause severe hemodynamic compromise and arrhythmic death.
Patients who develop secondary pulmonary hypertension (Eisenmenger syndrome) despite attempted correction of the anatomic defects have a very poor prognosis. The terminal event may be bradycardia or VT progressing to VF.
Acquired childhood diseases associated with VF include Kawasaki syndrome, DCM, and myocarditis. Causes of VF in patients with previously unrecognized structural heart disease include HCM, congenital coronary artery abnormalities, and ARVD.
Paroxysmal VF or short-coupled torsades
Paroxysmal familial VF can be caused by mutations in either the SCN5A or DPP6 gene. Mutations in the SCN5A gene are also associated with VF during MI. [32]
Idiopathic VF and VT
Idiopathic VF
Idiopathic VF is triggered by ventricular premature beats that may originate in the distal Purkinje conducting system, LV septum, anterior RV, or RV outflow tract (RVOT). Early repolarization, or J wave (elevation at the junction of between the QRS complex and ST-segment), has been identified in patients with idiopathic VF and has been associated with mutations in a variety of ion channel genes. [33] Catheter ablation that targets triggers of ventricular premature beats can provide long-term freedom from recurrence of idiopathic VF. [34]
Idiopathic VT
Although RVOT tachycardia is responsible for 70%-80% of idiopathic VTs, it is a rare cause of VF. Idiopathic VT is generally associated with a benign prognosis.
RVOT tachycardia has a left bundle-branch block/inferior or right-axis morphology on ECG. Idiopathic VTs that originate from the LV outflow tract (LVOT), aortic root, or LV septum are less common. However, with newer techniques of mapping and ablation, more PVC and VT foci are being mapped to the LVOT and aortic cusp area. Many patients with previous failed ablation for RVOT PVCs have undergone successful mapping and ablation in the LVOT or aortic cusp region. [35]
RVOT tachycardia is believed to be adrenergic-receptor mediated, because exogenous and endogenous adenosine can terminate the arrhythmia. Maneuvers that increase endogenous acetylcholine may also terminate the arrhythmia.
Symptoms typical of RVOT tachycardia include palpitations, presyncope, or syncope, often occurring during or after exercise or emotional stress, but VT can also occur at rest. VF has been reported in patients with RVOT who have PVCs with short coupling intervals (in contrast to the long coupling intervals seen in patients with benign forms of RVOT tachycardia). [36]
Treatment of RVOT is based on the symptomatic frequency and severity. Beta blockers are first-line therapy. Patients with symptoms not relieved by medical therapy are best treated with radiofrequency catheter ablation. Successful ablation is reported in 83%-100% of cases.
The next most common form of idiopathic VT arises from the fascicles of the LV, notably the left posterior fascicle. These patients present with an ECG pattern during VT showing a right bundle-branch block and superior axis. The initial drug of choice is a calcium channel blocker. Catheter ablation is very effective. Other, less common forms of idiopathic VT originate from the LV papillary muscles or from the crux of the heart (the region at the base of the ventricle nearest the atrioventricular [AV] node). [37]
Pulmonary embolism
Pulmonary embolism is a frequent cause of sudden death in at-risk people. Risk factors include a previous personal or family history of deep venous thromboembolism (DVT), malignancy, hypercoagulable states, and recent mechanical trauma such as hip or knee surgery. Patients with pulmonary embolism can develop fatal ventricular arrhythmias (eg, VF) due to hemodynamic collapse and/or severe hypoxia.
Aortic dissection
Aortic dissection or aneurysmal rupture is a rather uncommon, but significant, cause of out-of-hospital cardiac arrest. Predisposing factors for aortic dissection include genetic deficiencies of collagen such as Marfan syndrome, Ehlers-Danlos syndrome, and aortic cystic medial necrosis. VF may be an observed finding at the scene of an aortic aneurysmal rupture, or the sudden death from the hemodynamic collapse may be presumed to reflect associated VF without the presence of that rhythm.
Electronic control devices
Whether electronic control devices (eg, TASERs) can trigger VF has been studied in animal models, with contradictory results. [38] However, Zipes reported human cases in which TASER stimulation apparently caused cardiac electrical capture and provoked cardiac arrest from VT/VF. [39]
Nonstructural abnormalities
Nonstructural abnormalities generally are a group of abnormalities in which patients have no apparent structural heart disease but have a primary EP abnormality that predisposes them to VT or VF. [40] Some imaging techniques have detected abnormal sympathetic neural function in these patients. An ECG can provide clues to the diagnosis; consider a familial component to these conditions.
Causes of VF in patients with previously unrecognized, nonstructural heart disease include the following:
-
LQTS
-
CPVT
-
WPW syndrome
-
Primary VT and VF
-
Primary pulmonary hypertension
-
Commotio cordis (traumatic blow to the chest wall causing VT/VF)
-
Brugada syndrome: It is possible that some patients with what is thought to be primary VF may have Brugada syndrome; VF in these patients usually has no preceding symptoms; the prognosis is unfavorable, and the recurrence rate is as high as 33%.
Congenital long QT syndrome
Congenital LQTS results from abnormalities of channel proteins in the cardiac membrane. The most common forms involve loss of function of the potassium channel. Other forms may involve the sodium or calcium ion channels.
Prolongation of the QT interval is seen in the following genetic disorders:
-
Idiopathic LQTS
-
Romano-Ward syndrome
-
Jervell and Lange-Nielsen syndrome (JLNS)
-
Andersen-Tawil syndrome
-
Timothy syndrome
Current cardiologic practice, however, is moving away from eponyms and instead toward denoting LQTS by numbered type, based on identified underlying mutations. For example, long QT 1 is caused by mutations in the KCNQ1 gene; it is seen in Romano-Ward syndrome and JLNS.
The clinical course of LQTS is quite variable, with some patients remaining asymptomatic and others developing torsade de pointes with syncope and sudden death. In 30% of cases, the syndrome is identified during an evaluation for syncope or aborted sudden death.
Patients at high risk for VF include those with deafness and first-degree relatives of patients with VF. VF in these patients is associated with emotional extremes, auditory auras or stimulation, and vigorous physical activity. Symptoms usually begin in childhood or adolescence. Schwartz et al have suggested diagnostic criteria for LQTS in the absence of genetic testing (see Table 1, below). [41]
Table 1. Long QT syndrome diagnostic criteria (Open Table in a new window)
Category |
Criteria |
Points |
|
Electrocardiographic Findings |
Corrected QT interval |
≥480 ms |
3 |
460-479 ms |
2 |
||
450-459 ms (in males) |
1 |
||
Torsade de pointes |
2 |
||
T wave alternans |
1 |
||
Notched T waves in three leads |
1 |
||
Low heart rate for age (resting rate below second percentile |
0.5 |
||
Clinical History |
Syncope |
With stress |
2 |
Without stress |
1 |
||
Congenital deafness |
0.5 |
||
Family History |
Family members with definite long QT syndrome |
1 |
|
Unexplained sudden cardiac death before age 30 years in immediate family members without definite long QT syndrome |
0.5 |
||
Adapted from Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993 Aug;88(2):782-4. PMID: 8339437 [41] Scoring:
|
|||
The absence of a long QT interval on a single resting ECG does not exclude the diagnosis of LQTS. For example, it is possible that the patient may have incomplete penetrance that could be accentuated by drugs or metabolic conditions.
Treatment for LQTS includes beta blockers, high thoracic left sympathectomy, and ICDs. [42] For more information, see the Medscape Drugs and Diseases article Long QT Syndrome.
Idiopathic long QT syndrome
Idiopathic LQTS is characterized clinically by a propensity to develop malignant ventricular arrhythmias. It is a rare familial disorder.
Romano-Ward syndrome
Romano-Ward syndrome describes a family of nonsyndromic LQTS. It is characterized by prolongation of the QT interval, as well as T-wave abnormalities and polymorphic VT. Patients with this disease are predisposed to events of polymorphic VT, which can be self-limited, resulting in syncope. It can also transition into VF and can cause SCD.
Romano-Ward syndrome is inherited in an autosomal dominant fashion, with a penetrance of approximately 50%. Mutations in the KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 genes are known to be causative, and these five genes together are responsible for virtually 100% of cases of Romano-Ward syndrome.
Jervell and Lange-Nielsen syndrome
JLNS is characterized by congenital sensorineural deafness and a prolonged QT interval. Cardiac events tend to occur at a young age. JLNS is caused by mutations in the KCNQ1 or KCNE1 gene and has an autosomal recessive pattern of inheritance. Persons who are heterozygous for mutations in these genes may be asymptomatic or may manifest Romano-Ward syndrome, but they will have normal hearing.
Andersen-Tawil syndrome
Andersen-Tawil syndrome is chiefly characterized by the triad of periodic flaccid paralysis, prolonged QT interval, and dysmorphic facies. Patients with Andersen-Tawil syndrome have low-set ears, hypertelorism, micrognathia, syndactyly, and short stature. [31] They may also have mild learning disability.
This syndrome is caused by mutation in the KCNJ2 gene, which encodes the ik1 channel and is inherited in an autosomal dominant fashion. About half of affected patients have a de novo mutation.
Timothy syndrome
Timothy syndrome is characterized by the presence of a prolonged QT interval and cutaneous syndactyly. The syndactyly may be unilateral or bilateral and may be of variable severity. Other possible manifestations include cardiac defects, characteristic facial features, and neurologic problems.
Timothy syndrome is caused by mutation in the CACN1C gene, which encodes the α subunit of the calcium channel. It is usually the result of a de novo mutation but is transmitted in an autosomal dominant fashion. [43]
Acquired long QT syndrome
A number of antiarrhythmics (especially class Ia and class III) and other medications, electrolyte abnormalities, cerebrovascular diseases, and altered nutritional states cause QT prolongation and put patients at risk for torsade de pointes. This usually occurs when QT prolongation is associated with a slow heart rate and hypokalemia.
The QT interval is prolonged in up 32% of patients with intracranial hemorrhage (especially subarachnoid hemorrhage). Stroke or intracranial trauma can also result in QT interval prolongation. Lesions in the hypothalamus are thought to lead to this phenomenon. In rare cases, QT prolongation from subarachnoid hemorrhage results in torsades de pointes.
Electrolyte abnormalities that cause acquired LQTS include hypokalemia, hypomagnesemia, and hypocalcemia. Such abnormalities may result from nutritional deficiencies associated with modified starvation diets and severe weight-loss programs. Altered autonomic status (eg, diabetic neuropathy) can cause LQTS. Rarely, hypothyroidism may result in QT interval prolongation.
Class Ia antiarrhythmic drugs that cause acquired LQTS include quinidine, disopyramide, and procainamide. Class III antiarrhythmic drugs that cause acquired LQTS include the following:
-
Sotalol
-
Amiodarone
-
Dofetilide
-
Ibutilide
Other drugs that can cause acquired LQTS include the following:
-
Tricyclic and tetracyclic antidepressants
-
Phenothiazines
-
Haloperidol
-
Antibiotics (eg, intravenous erythromycin, sulfamethoxazole/trimethoprim)
-
Chemotherapeutics (eg, pentamidine, anthracycline)
-
Serotonin antagonists (eg, ketanserin, zimeldine)
-
Organophosphorus insecticides
A comprehensive list of drugs that can prolong the QT interval is available at http://www.crediblemeds.org.
Catecholaminergic polymorphic VT
CPVT can be triggered by emotional stress or exercise and can also be induced by catecholamine administration. Patients may present with syncope, or SCD may occur if polymorphic VT degrades into VF. Results of physical examination or resting ECG are usually normal.
CPVT may be caused by mutations in the RYR2 gene (autosomal dominant) or the CASQ2 gene (autosomal recessive). [44] An additional locus has been mapped to chromosome 7p22-p14. Symptoms, including sudden death, usually appear in childhood or among young adults. Most cases of CPVT respond to beta-blocker therapy. Flecainide has also been found to be beneficial. [45] Invasive treatments include left cervical sympathectomy, ICD placement, or both.
Wolff-Parkinson-White syndrome
WPW syndrome is a rare cause of sudden death. The presence of multiple accessory pathways, posteroseptal accessory pathways, and a preexcited R-R interval of less than 220 msec during atrial fibrillation is associated with higher risk for VF. [46]
Most cases of VF in patients with WPW syndrome are induced by atrial fibrillation with a rapid ventricular response over the accessory pathway (see the image below). In a study by Klein et al of 31 patients with VF and WPW syndrome, a history of atrial fibrillation or reciprocating tachycardia was an important predisposing factor. [46]
 Ventricular fibrillation appeared during rapid atrial fibrillation in a patient with Wolff-Parkinson-White syndrome.
Ventricular fibrillation appeared during rapid atrial fibrillation in a patient with Wolff-Parkinson-White syndrome.
Mutation in the PRKAG2 gene can cause WPW. The syndrome is likely inherited in an autosomal dominant manner with reduced penetrance. It is not known what percentage of patients with WPW has a mutation in this gene.
Treatment of WPW should be individualized for each patient and is based in part on risk assessment. Risk assessment can be noninvasive, but symptomatic patients and those with sustained preexcitation during a treadmill exercise stress test require invasive risk stratification via an EP study (EPS.
In high-risk patients (ie, those with multiple pathways, a history of sustained reciprocating tachycardia, or short preexcited RR interval during atrial fibrillation [< 220 msec]), preventing the occurrence of arrhythmias is possible by interrupting the anomalous pathway with catheter ablation. Autonomic tone can alter conduction over a bypass tract, and complete assessment of the “shortest preexcited RR interval” includes assessment with the infusion of isoproterenol. [47]
Atrial fibrillation in patients with WPW can be associated with rapid ventricular rates due to rapid conduction over an accessory pathway with a short refractory period. Pure AV nodal blocking drugs (beta blockers, calcium channel blockers) should not be used alone, as they will block the AV node without affecting the accessory pathway; this can increase preferential conduction over the pathway, leading to faster ventricular rates.
Drugs such as ibutilide, procainamide, and amiodarone are useful, as they prolong the anterograde refractory period of the pathway and hence slow the conduction over the pathway. Electrical cardioversion is the preferred treatment for patients with hemodynamic instability.
Brugada syndrome
Brugada syndrome was first described in 1992 by Brugada and Brugada. [48] It is characterized by a specific ECG pattern of right bundle-branch block and ST-segment elevation in leads V1 and V2 without any structural abnormality of the heart. [49]
Brugada syndrome can be caused by mutations in many genes. The principal gene associated with the syndrome is SCN5A, over 400 mutations of which have been described. [50, 51] Other genes known to cause the syndrome include GPD1L, CACNA1C, CACNB2, SCN1B, KCNE3, SCN3B, and HCN4. Brugada syndrome is inherited in an autosomal dominant fashion.
Patients with this syndrome are at high risk for VF. In equivocal cases, intravenous (IV) procainamide will augment the Brugada ECG pattern. [52] In an early follow-up study of 63 patients with the syndrome, asymptomatic patients were found to have the same risk for arrhythmia as patients who had an episode of aborted sudden death. [53] In this study, treatment with amiodarone, beta blockers, or both did not confer a lower risk of death, whereas the patients with ICDs had no deaths due to arrhythmia. Thus, placement of an ICD is considered the treatment of choice for Brugada syndrome. [53]
Larger, subsequent studies have emphasized a much lower risk for SCD in asymptomatic patients with an episodic Brugada pattern. [54] Other therapeutic options include IV isoproterenol for management of VF storm and oral quinidine as outpatient therapy for avoiding ICD shocks. [55] Other important aspects of therapy include prompt treatment of fever and avoidance of drugs that cause sodium channel blockade.
Epidemiology
United States and international data
Many episodes of ventricular fibrillation (VF) are unwitnessed, making it difficult to assess an exact incidence. Of the approximately 300,000 cases of SCD that occur each year in the United States, up to one third are attributed to VF. [56] This represents an incidence of 0.08%-0.16% per year in the adult population, accounting for more deaths than from lung cancer, breast cancer, or acquired immunodeficiency syndrome (AIDS). In the pediatric and adolescent age groups, VF occurs with an annual incidence of 1.3-8.5 cases per 100,000 persons, accounting for approximately 5% of all deaths in this group.
VF is often the first expression of coronary artery disease (CAD) and is responsible for approximately 50% of deaths from CAD. VF often occurs within the first hour after the onset of an acute myocardial infarction (MI) or acute coronary syndrome (ACS).
In several population-based studies, although the incidence of out-of-hospital cardiac arrest in the United States has been noted as declining in the past 2 decades, the proportion of sudden deaths from VF in patients with CAD has not changed. A high incidence of VF occurs among certain population subgroups (eg, patients with chronic heart failure with ejection fraction < 30%, patients in the convalescent phase after MI, patients who survived cardiac arrest); however, only a small percentage of total VF events occur in these patients, because the population size is small relative to lower-risk groups.
Survivors of a major cardiovascular event have an increased risk for VF in the first 6-24 months after the event. Up to 30% of survivors of cardiac arrest may experience recurrent VF in the first year afterward.
The frequency of VF in other industrialized Western nations is similar to that in the United States. [57] The incidence of VF in other countries varies as a reflection of CAD prevalence in those populations. The trend toward an increasing frequency of VF events in developing nations is thought to reflect a change in dietary and lifestyle habits.
Cardiovascular events, including sudden cardiac death (SCD) from VF (but not asystole), most frequently occur in the morning and may be related to increased platelet aggregability. A spike in the number of SCDs also appears to occur during the winter months.
Race-, sex-, and age-related demographics
Most data are inconclusive regarding racial differences and the incidence of VF. Some studies suggest that a greater proportion of coronary deaths have been sudden in black individuals than in white individuals. [58] In a report by Gillum on SCD from 1980-1985 data, the percentage of CAD deaths occurring out of the hospital and in emergency departments was higher in black patients than in white patients. [59]
The incidence of VF is higher in men than in women (3:1). [60] This ratio generally reflects the higher incidence of CAD in men. Although the mechanism of MI tends to differ by sex—coronary plaque rupture in men and plaque erosion in women—it is unclear whether this difference accounts for the male predominance of VF.
The incidence of VF parallels the prevalence of CAD, with the peak rate of VF occurring in people aged 45-75 years. However, the proportion of sudden deaths from CAD decreases with age. In the Framingham Heart Study, the proportion of sudden CAD deaths was 62% in men aged 45-54 years, decreasing to 58% in men aged 55-64 years and to 42% in men aged 65-74 years. [61] According to Kuller, 31% of deaths are sudden in people aged 20-29 years. [62]
Prognosis
The chances of survival from an index ventricular fibrillation (VF) event depend on bystander cardiopulmonary resuscitation (CPR), rapid availability or arrival of personnel and apparatus for defibrillation and advanced life support, and transport to a hospital. Although patients with nontraumatic cardiac arrest are more likely to be successfully resuscitated from VF than from any other arrhythmia, success is highly time dependent. The probability of success generally declines at a rate of 2%-10% per minute.
Early defibrillation often makes the difference between long-term disability and functional recovery. Placement of automated external defibrillators (AEDs) throughout communities and training of the public in their use has the potential to improve outcomes from sudden cardiac death (SCD). [63]
In patients presenting to an emergency department (ED) after a witnessed episode of VF, the prognosis for morbidity and mortality can be determined by calculating the cardiac arrest score, developed by McCullough and Thompson. [64] This score is based on systolic blood pressure, time from loss of consciousness to return of spontaneous circulation, and neurologic responsiveness. (See Presentation for details.)
Even under ideal circumstances, however, only an estimated 20% of persons who have out-of-hospital cardiac arrest survive to hospital discharge. In a study of out-of-hospital cardiac arrest survival in New York City, only 1.4% of patients survived to hospital discharge. [65] However, studies in suburban and rural areas have indicated survival rates of up to 35%. [66]
Routine coronary angiography, with percutaneous coronary intervention (PCI), if indicated, along with mild therapeutic hypothermia (core temperature of 32°-34°C for 24 hours), may favorably alter the prognosis of resuscitated patients with stable hemodynamics after out-of-hospital cardiac arrest. [67] In a retrospective study of cardiac arrest survivors, 65.6% of patients who underwent early coronary angiography survived to hospital discharge, compared with 48.6% of those who did not receive coronary angiography. [68]
A major adverse outcome from VF episodes is anoxic encephalopathy, which occurs in 30%-80% of patients. A study conducted in Minnesota on all adult survivors of out-of-hospital VF-related cardiac arrest from 1990-2008 found that long-term survivors had long-term memory deficits. [69] A 2017 report that evaluated 2009-2013 data from the Pan-Asian Resuscitation Outcomes Study (PAROS) registry to determine characteristics and outcomes of 3244 young adults (aged 16-35 years) who suffered out-of-hospital cardiac arrest found that factors associated with favorable neurologic outcomes included first arrest rhythms of VF/VT/unknown shockable rhythm, cardiac etiology, bystander-witnessed arrest, and bystander CPR. [70] However, traumatic out-of-hospital cardiac arrest had a poor prognosis.
-
This image shows an epsilon wave on the electrocardiogram of a patient with arrhythmogenic right ventricular dysplasia (ARVD).
-
Ventricular fibrillation appeared during rapid atrial fibrillation in a patient with Wolff-Parkinson-White syndrome.
-
This image reveals ventricular fibrillation in a patient with a left ventricular assist device (LVAD).
-
Position of the paddle electrodes during defibrillation/cardioversion, position of the heart, and flow of intrathoracic energy during delivery of the electric shock are shown.
Tables
Category |
Criteria |
Points |
|
Electrocardiographic Findings |
Corrected QT interval |
≥480 ms |
3 |
460-479 ms |
2 |
||
450-459 ms (in males) |
1 |
||
Torsade de pointes |
2 |
||
T wave alternans |
1 |
||
Notched T waves in three leads |
1 |
||
Low heart rate for age (resting rate below second percentile |
0.5 |
||
Clinical History |
Syncope |
With stress |
2 |
Without stress |
1 |
||
Congenital deafness |
0.5 |
||
Family History |
Family members with definite long QT syndrome |
1 |
|
Unexplained sudden cardiac death before age 30 years in immediate family members without definite long QT syndrome |
0.5 |
||
Adapted from Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993 Aug;88(2):782-4. PMID: 8339437 [41] Scoring:
|
|||
Cardiac Arrest Score |
In-Hospital Mortality Rate (%) |
Neurologic Recovery (%) |
0 |
90 |
3 |
1 |
71 |
17 |
2 |
42 |
57 |
3 |
18 |
89 |





