Practice Essentials
Beta thalassemia syndromes are a group of hereditary disorders characterized by a genetic deficiency in the synthesis of beta-globin chains. In the homozygous state, beta thalassemia (ie, thalassemia major) causes severe, transfusion-dependent anemia. In the heterozygous state, the beta thalassemia trait (ie, thalassemia minor) causes mild to moderate microcytic anemia. (See Etiology.)
Patients in whom the clinical severity of the disease lies between that of thalassemia major and thalassemia minor are categorized as having thalassemia intermedia. Several different genotypes are associated with thalassemia intermedia.
Hemoglobin (Hb) E, a common Hb variant found in Southeast Asia, is associated with a beta thalassemia phenotype, and this variant is included in the beta thalassemia category of diseases.
Patients with thalassemia minor usually do not require any specific treatment. Treatment for patients with thalassemia major includes long-term transfusion therapy, iron chelation, splenectomy, allogeneic hematopoietic stem cell transplantation, gene therapy, and supportive measures. See Treatment.
Complications associated with beta thalassemia
Complications associated with beta thalassemia, aside from the aforementioned anemia, are as follows (see Prognosis, Presentation, Workup, Treatment, and Medication):
-
Extramedullary hematopoiesis
-
Asplenia secondary to splenectomy
-
Medical complications from long-term transfusional therapy - Iron overload and transfusion-associated infections (eg, hepatitis); iron overload cardiomyopathy accounts for the majority of deaths in thalassemia patients [1]
-
Increased risk for infections resulting from asplenia (eg, encapsulated organisms such as pneumococcus) or from iron overload (eg, Yersinia species)
-
Cholelithiasis (eg, bilirubin stones)
Etiology
Beta-globin gene mutations
Mutations in globin genes cause thalassemias. (Mutations in alpha-globin genes cause alpha thalassemia.) Beta thalassemia is due to mutations in one or both of the beta-globin genes that result in impaired synthesis of the beta-globin protein component of Hb, and subsequently in anemia. [2, 3, 4]
More than 350 beta-globin gene mutations have been identified in patients with beta thalassemia; this underlies the wide genotypic and phenotypic variability of the disease. Worldwide, however, only 20 mutations account for more than 80% of cases, because of geographical clustering of populations with a few common mutations. [5]
Beta thalassemia is inherited as an autosomal recessive disorder. The defect can be a complete absence of the beta-globin protein (ie, beta-zero thalassemia) or a severely reduced synthesis of the beta-globin protein (ie, beta-plus thalassemia). (See the image below.)
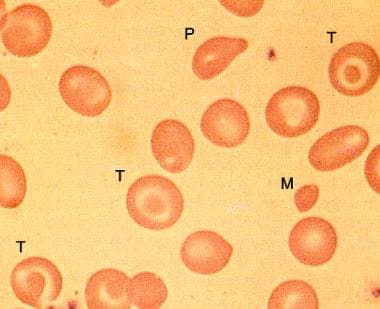 Peripheral smear in beta-zero thalassemia minor showing microcytes (M), target cells (T), and poikilocytes.
Peripheral smear in beta-zero thalassemia minor showing microcytes (M), target cells (T), and poikilocytes.
The genetic defect usually is a missense or nonsense mutation in the beta-globin gene. Occasional defects involving deletions of the beta-globin gene and surrounding chromosomal regions also have been reported.
In beta thalassemia minor (ie, beta thalassemia trait or heterozygous carrier-type), one of the beta-globin genes is defective, resulting in an approximately 50% decrease in the synthesis of the beta-globin protein.
In beta thalassemia intermedia the production of beta-globin chains is variably decreased. This is generally attributed to the presence of two beta-globin gene mutations, but because of the heterogeneity of beta-globin mutations and the large number of possiblre disease modifiers, beta thalassemia intermedia is often defined clinically: these patients require transfusions only sporadically, if at all.
In beta thalassemia major (ie, homozygous beta thalassemia), the production of the beta-globin chains is severely impaired because both beta-globin genes are mutated. The severe imbalance of globin chain synthesis (alpha >> beta) results in ineffective erythropoiesis and severe microcytic hypochromic anemia. (See the image below.) The excess unpaired alpha-globin chains aggregate to form precipitates that damage red cell membranes, resulting in intravascular hemolysis. Premature destruction of erythroid precursors results in intramedullary death and ineffective erythropoiesis. The profound anemia typically is associated with erythroid hyperplasia and extramedullary hematopoiesis.
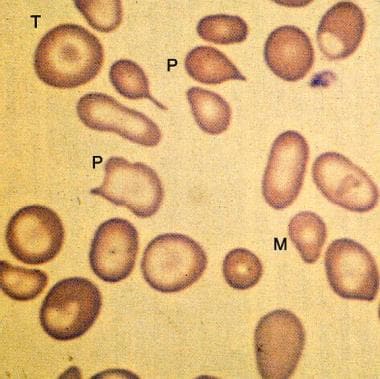 Peripheral smear from a patient with beta-zero thalassemia major showing more marked microcytosis (M) and anisopoikilocytosis (P) than in thalassemia minor. Target cells (T) and hypochromia are prominent.
Peripheral smear from a patient with beta-zero thalassemia major showing more marked microcytosis (M) and anisopoikilocytosis (P) than in thalassemia minor. Target cells (T) and hypochromia are prominent.
Beta thalassemia major is also known as Cooley anemia. The eponym honors Dr. Thomas Denton Cooley, who described what he called erythroblastic anemia in 1925, based on studies in Michigan children of Italian and Greek heritage. [6]
Although beta thalassemia is caused by mutations in the beta-globin gene (which is located on chromosome 11), many additional factors influence the clinical manifestations of the disease. That is, the same mutations may have different clinical manifestations in different patients. The factors below are known to influence the clinical phenotype.
Intracellular fetal Hb concentrations
The level of expression of fetal Hb (ie, the expression level of the gamma-globin gene) in red blood cells determines, in part, the severity of the disease. Patients with high fetal Hb have milder disease.
Coinheritance of alpha thalassemia
Patients with coinheritance of alpha thalassemia have a milder clinical course because they have a less severe alpha-beta chain imbalance.
Coexistence of sickle cell trait
The coexistence of sickle cell trait and beta thalassemia is a major and symptomatic hemoglobinopathy with most of the symptoms and complications of sickle cell disease. Unlike sickle cell trait, in which most Hb-on-Hb electrophoresis is Hb A (AS), S is the dominant Hb (SA) and usually constitutes about 60% or more of the circulating Hb, depending on the transfusion status of the patient and the nature of the coexisting beta-thalassemia mutation (ie, beta-zero vs beta-plus).
Epidemiology
Occurrence in the United States
The frequency of beta thalassemia varies widely, depending on the ethnic population. The disease is reported most commonly in Mediterranean, African, and Southeast Asian populations.
International occurrence
The disease is found most commonly in the Mediterranean region, Africa, and Southeast Asia, presumably as an adaptive association to endemic malaria. The prevalence may be as high as 10% in these areas.
Race-related demographics
Beta thalassemia genes are reported throughout the world, although more frequently in Mediterranean, African, and Southeast Asian populations. Patients of Mediterranean extraction are more likely than Africans to be anemic with thalassemia trait, because they tend to have beta-zero thalassemia rather than beta-plus thalassemia.
The genetic defect in Mediterranean populations is most commonly either (1) a mutation creating an abnormal splicing site or (2) a mutation creating a premature translation termination codon. Southeast Asian populations also have a significant prevalence of Hb E and alpha thalassemia. African populations more commonly have genetic defects leading to alpha thalassemia.
Age-related demographics
The manifestations of the disease may not be apparent until a complete switch from fetal to adult Hb synthesis occurs. This switch typically is completed by the sixth month after birth.
Prognosis
Individuals with thalassemia minor (thalassemia trait) usually have mild, asymptomatic microcytic anemia. This state does not result in mortality or significant morbidity.
The prognosis of patients with thalassemia major is highly dependent on the patient's adherence to long-term treatment programs, namely the hypertransfusion program and lifelong iron chelation. Allogeneic bone marrow transplantation may be curative.
Morbidity and mortality
The major causes of morbidity and mortality in beta thalassemia are anemia and iron overload. The severe anemia resulting from this disease, if untreated, can result in high-output cardiac failure; the intramedullary erythroid expansion may result in associated skeletal changes such as cortical bone thinning. The long-term increase in red-cell turnover causes hyperbilirubinemia and bilirubin-containing gallstones.
Increased iron deposition resulting from lifelong transfusions and enhanced iron absorption results in secondary iron overload. This overload causes clinical problems similar to those observed with primary hemochromatosis (eg, endocrine dysfunction, liver dysfunction, cardiac dysfunction).
A broad spectrum of neurological complications has also been reported in beta thalassemia complications, although most were subclinical. These have included the following [7] :
-
Cognitive impairment
-
Abnormal findings on evoked potentials
-
Cerebrovascular disease
-
Peripheral neuropathy
Patient Education
Educate patients with thalassemia minor about the genetic (hereditary) nature of their disease, and inform them that their immediate family members (ie, parents, siblings, children) may be affected. The presence of beta-thalassemia minor in both parents implies that there is about a one fourth chance that a child will have thalassemia major. Careful genetic counseling is also appropriate for patients in whom one parent has beta-thalassemia minor and the other parent has some form of beta-globin–related disease, such as sickle cell carriage.
Inform patients with thalassemia minor that they do not have iron deficiency and that iron supplementation will not improve their anemia.
For patient education information, see the Thalassemia Directory.
-
Peripheral smear in beta-zero thalassemia minor showing microcytes (M), target cells (T), and poikilocytes.
-
Peripheral smear from a patient with beta-zero thalassemia major showing more marked microcytosis (M) and anisopoikilocytosis (P) than in thalassemia minor. Target cells (T) and hypochromia are prominent.








