Practice Essentials
Bartter syndrome, originally described by Bartter and colleagues in 1962, [1] represents a set of closely related, autosomal recessive renal tubular disorders characterized by hypokalemia, hypochloremia, metabolic alkalosis, and hyperreninemia with normal blood pressure. [2, 3] The underlying kidney abnormality results in excessive urinary losses of sodium, chloride, and potassium.
Bartter syndrome has traditionally been classified into three main clinical variants, as follows:
-
Neonatal (or antenatal) Bartter syndrome
-
Classic Bartter syndrome
-
Gitelman syndrome
Advances in molecular diagnostics have revealed that Bartter syndrome results from mutations in numerous genes that affect the function of ion channels and transporters that normally mediate transepithelial salt reabsorption in the distal nephron segments (see the image below). Hundreds of mutations have been identified to date. Such advances may result in the development of new therapies. [4] (See Pathophysiology and Etiology.)
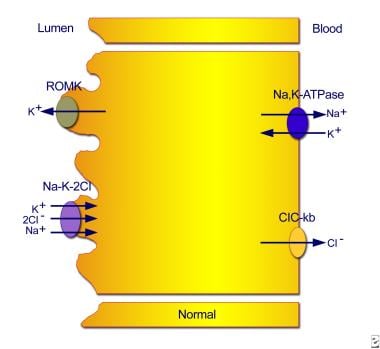 Normal transport mechanisms in the thick ascending limb of the loop of Henle. Reabsorption of sodium chloride is achieved with the sodium chloride/potassium chloride cotransporter, which is driven by the low intracellular concentrations of sodium, chloride, and potassium. Low concentrations are maintained by the basolateral sodium pump (sodium-potassium adenosine triphosphatase), the basolateral chloride channel (ClC-kb), and the apical potassium channel (ROMK).
Normal transport mechanisms in the thick ascending limb of the loop of Henle. Reabsorption of sodium chloride is achieved with the sodium chloride/potassium chloride cotransporter, which is driven by the low intracellular concentrations of sodium, chloride, and potassium. Low concentrations are maintained by the basolateral sodium pump (sodium-potassium adenosine triphosphatase), the basolateral chloride channel (ClC-kb), and the apical potassium channel (ROMK).
More recently, other classification systems for Bartter syndrome have been developed. Seyberth proposed a classification of Bartter syndrome that takes into account the three main anatomic and pathophysiologic disturbances that lead to the salt-losing tubulopathy. The classification is as follows (see Etiology, Presentation, and Workup) [5] :
-
Classic Bartter syndrome and Gitelman syndrome - The first type involves dysfunction in the thick ascending limb of the loop of Henle (TALH) or distal convoluted tubule (DCT) that leads to hypokalemia; this condition takes the form of either classic Bartter syndrome (caused by mutations in the CLCNKB gene) or Gitelman syndrome (caused by mutations in the NCCT gene). In addition, a mutation in the basolateral calcium sensing receptor has been identified as causing milder symptoms of classic Bartter syndrome. [6]
-
Neonatal (or antenatal) Bartter syndrome - The second type involves polyuric loop dysfunction that is more severe; this form of Bartter syndrome is characterized by defects in the NKCC2 and ROMK genes
-
Neonatal (or antenatal) Bartter syndrome with sensorineural deafness - The third type involves the most severe combined loop and distal convoluted tubule dysfunction; it is caused by defects in the chloride channel genes CLCNKB and CLCNKA or their beta subunit BSND.
A widely used system classifies Bartter syndrome on the basis of the underlying genetics, as follows [7] :
-
Type 1 – Antenatal Bartter syndrome: Results from mutations in SLC12A1, the sodium-chloride-potassium cotransporter gene
-
Type 2 – Antenatal/neonatal Bartter syndrome: Results from mutations in the ROMK gene
-
Type 3 – Classic Bartter syndrome: caused by mutations of the chloride voltage-gated channel Kb gene ( CLCNKB), which encodes the kidney-specific basolateral chloride channel (ClC-Kb) involved in sodium chloride reabsorption in the renal tubule [8]
-
Type 4 – Bartter syndrome with sensorineural deafness: Results from loss-of-function mutations in BSND, which encodes an essential beta subunit for chloride channels
-
Type 5 – Gitelman syndrome: Results from mutations in SLC12A3, the sodium-chloride cotransporter
Most recently, an international team of researchers has identified an X-linked disorder characterized by polyhydramnios with prematurity and a severe but transient form of antenatal Bartter syndrome. The disorder results from mutations in MAGED2, a gene on the X chromosome that encodes melanoma-associated antigen D2 (MAGE-D2), which is essential for fetal renal salt reabsorption, amniotic fluid homeostasis, and the maintenance of pregnancy. In their study of 13 infants, four died perinatally and 11 survived; in the survivors, all symptoms disappeared spontaneously during follow-up. [9] In a French cohort, MAGED2 mutations accounted for 9% of antenatal Bartter syndrome and 38% of patients without other characterized mutations. [10]
Pathophysiology
Bartter and Gitelman syndromes are renal tubular salt-wasting disorders in which the kidneys cannot reabsorb chloride in the loop of Henle (TALH) or distal convoluted tubule (DCT), depending on the mutation.
Chloride is passively absorbed along most of the proximal tubule but is actively transported in the TALH and the DCT. Failure to reabsorb chloride results in a failure to reabsorb sodium and leads to excessive sodium and chloride (salt) delivery to the distal tubules, with consequent excessive salt and water loss from the body.
Other pathophysiologic abnormalities result from excessive salt and water loss. The renin-angiotensin-aldosterone system (RAAS) is a feedback system activated with volume depletion. Long-term stimulation may lead to hyperplasia of the juxtaglomerular complex.
Angiotensin II (ANG II) is directly vasoconstrictive, increasing systemic and renal arteriolar constriction, which helps to prevent systemic hypotension. It directly increases proximal tubular sodium reabsorption.
ANG II–induced renal vasoconstriction, along with potassium deficiency, produces a counterregulatory rise in vasodilating prostaglandin E (PGE) levels. High PGE levels are associated with growth inhibition in children.
High levels of aldosterone also enhance potassium and hydrogen exchange for sodium. Excessive intracellular hydrogen ion accumulation is associated with hypokalemia and intracellular renal tubule potassium depletion. This is because hydrogen is exchanged for potassium to maintain electrical neutrality. It may lead to intracellular citrate depletion, because the alkali salt is used to buffer the intracellular acid and then lowers urinary citrate excretion. Hypocitraturia is an independent risk factor for renal stone formation.
Excessive distal sodium delivery increases distal tubular sodium reabsorption and exchange with the electrically equivalent potassium or hydrogen ion. This, in turn, promotes hypokalemia, while lack of chloride reabsorption promotes inadequate exchange of bicarbonate for chloride, and the combined hypokalemia and excessive bicarbonate retention lead to metabolic alkalosis.
Persons with Bartter syndrome often have hypercalciuria. Normally, reabsorption of the negative chloride ions promotes a lumen-positive voltage, driving paracellular positive calcium and magnesium absorption. Continued reabsorption and secretion of the positive potassium ions into the lumen of the TALH also promotes reabsorption of the positive calcium ions through paracellular tight junctions. Dysfunction of the TALH chloride transporters prevents urine calcium reabsorption in the TALH. Excessive urine calcium excretion may be one factor in the nephrocalcinosis observed in these patients.
Calcium is usually reabsorbed in the DCT. Theoretically, chloride is reabsorbed through the thiazide-sensitive sodium chloride cotransporter and transported from the cell through a basolateral chloride channel, reducing intracellular chloride concentration. The net effect is increased activity of the voltage-dependent calcium channels and enhanced electrical gradient for calcium reabsorption from the lumen.
In Gitelman syndrome, dysfunction of the sodium chloride cotransporter (NCCT) leads to hypocalciuria and hypomagnesemia. In the last several years, the understanding of magnesium handling by the kidney has improved and advances in genetics have allowed the differentiation of a variety of magnesium-handling mutations.
While patients with the variants that make up Bartter syndrome may or may not have hypomagnesemia, this condition is pathognomonic for Gitelman syndrome. The mechanism of the impaired magnesium reabsorption is still unknown; studies in NCCT knockout mice demonstrate increased apoptosis of DCT cells, which would then lead to diminished reabsorptive surface area. [11]
Sensorineural deafness
The ClC-Kb channel is found in the basolateral membrane of the TALH, while the barttin subunits of ClC-Ka and ClC-Kb are found in the basolateral membrane of the marginal cells of the cochlear stria vascularis.
In the inner ear, an Na-K-2Cl pump, called NKCC1, on the basolateral membrane increases intracellular levels of sodium, potassium, and chloride. Potassium excretion across the apical membrane against a concentration gradient produces the driving force for the depolarizing influx of potassium through the ion channels of the sensory hair cells required for hearing. The sodium ion is excreted across the basolateral membrane by the Na-K-adenosine triphosphatase (ATPase) pump, and the ClC-K channels allow the chloride ion to exit to maintain electroneutrality.
Sensorineural deafness associated with type IV Bartter syndrome, a neonatal form of the disease (see Etiology), is due to defects in the barttin subunit of the ClC-Ka and CIC-Kb channels.
Mutations in only the ClC-Kb subunit, as occurs in type III Bartter syndrome, do not result in sensorineural deafness.
Etiology
Defects in either the sodium chloride/potassium chloride cotransporter or the potassium channel affect the transport of sodium, potassium, and chloride in the thick ascending limb of the loop of Henle (TALH). The result is the delivery of large volumes of urine with a high content of these ions to the distal segments of the renal tubule, where only some sodium is reabsorbed and potassium is secreted.
Familial and sporadic forms of Bartter and Gitelman syndromes exist. When inherited, these syndromes are passed on as autosomal recessive conditions.
Neonatal (type I and type II) Bartter syndrome
An autosomal recessive mode of inheritance is observed in some patients with neonatal Bartter syndrome, although many cases are sporadic.
Type I results from mutations in the sodium chloride/potassium chloride cotransporter gene (NKCC2; locus SLC12A1 on chromosome bands 15q15-21). (See the first image below.) Type II results from mutations in the ROMK gene (locus KCNJ1 on chromosome bands 11q24-25). (See the second image below.) Numerous mutations have been identified at those sites. [12, 13, 14]
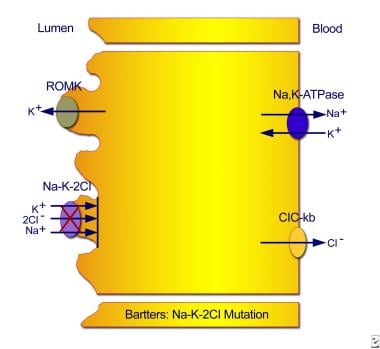 Type I neonatal Bartter syndrome. Mutations in the sodium chloride/potassium chloride cotransporter gene result in defective reabsorption of sodium, chloride, and potassium.
Type I neonatal Bartter syndrome. Mutations in the sodium chloride/potassium chloride cotransporter gene result in defective reabsorption of sodium, chloride, and potassium.
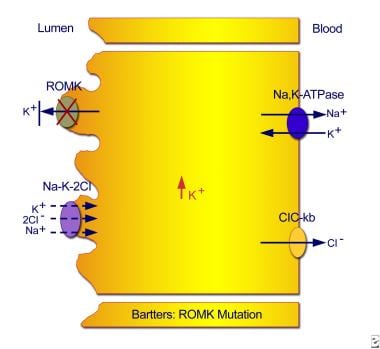 Type II neonatal Bartter syndrome. Mutations in the ROMK gene result in an inability to recycle potassium from the cell back into the tubular lumen, with resultant inhibition of the sodium chloride/potassium chloride cotransporter.
Type II neonatal Bartter syndrome. Mutations in the ROMK gene result in an inability to recycle potassium from the cell back into the tubular lumen, with resultant inhibition of the sodium chloride/potassium chloride cotransporter.
Classic (type III) Bartter syndrome
Some patients have an autosomal recessive mode of inheritance in classic Bartter syndrome, although many cases are sporadic.
In classic Bartter syndrome, the defect in sodium reabsorption appears to result from mutations in the chloride-channel gene (CLCNKB, on band 1p36). The consequent inability of chloride to exit the cell inhibits the sodium chloride/potassium chloride cotransporter. (See the image below.)
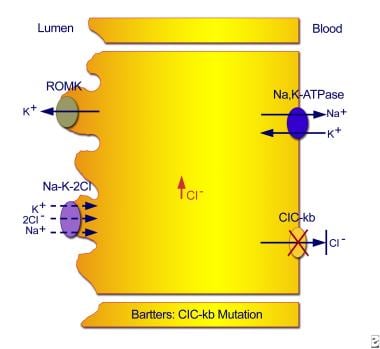 Classic Bartter syndrome. Mutations in the ClC-kb chloride channel lead to an inability of chloride to exit the cell, with resultant inhibition of the sodium chloride/potassium chloride cotransporter.
Classic Bartter syndrome. Mutations in the ClC-kb chloride channel lead to an inability of chloride to exit the cell, with resultant inhibition of the sodium chloride/potassium chloride cotransporter.
Increased delivery of sodium chloride to the distal sites of the nephron leads to salt wasting, polyuria, volume contraction, and stimulation of the renin-angiotensin-aldosterone axis. These effects, combined with biologic adaptations of downstream tubular segments, specifically the distal convoluted tubule (DCT) and the collecting duct, result in hypokalemic metabolic alkalosis. [15]
The hypokalemia, volume contraction, and elevated angiotensin levels increase intrarenal prostaglandin E2 (PGE2) synthesis, which contributes to a vicious cycle by inhibiting sodium chloride reabsorption in the TALH and further stimulating the renin-aldosterone axis.
Recently, a novel mutation in the CLCNKB gene was reported in three siblings with short stature and growth retardation. Genetic analysis showed a c.2 T>G/delta exone 1-19) compound heterozygous mutation seen in all three children. This was associated with low basal and stimulated growth hormone and IGF-1 levels. [16]
Type IV Bartter syndrome
Studies have identified a novel type IV Bartter syndrome. [17, 18, 19] This is a type of neonatal Bartter syndrome associated with sensorineural deafness and has been shown to be caused by mutations in the BSND gene. [18, 20, 21] BSND encodes barttin, an essential beta subunit that is required for the trafficking of the chloride channel ClC-K (ClC-Ka and ClC-Kb) to the plasma membrane in the TALH and the marginal cells in the scala media of the inner ear that secrete potassium ion ̶ rich endolymph. [17] Thus, loss-of-function mutations in barttin cause Bartter syndrome with sensorineural deafness.
In contrast to other Bartter types, the underlying genetic defect in type IV is not directly in an ion-transporting protein. The defect instead indirectly interferes with the barttin-dependent insertion in the plasma membrane of chloride channel subunits ClC-Ka and ClC-Kb. [22]
Type IVb Bartter syndrome
Type IVb Bartter syndrome is a recently renamed form. It is associated with sensorineural deafness but is not caused by mutations in the BSND gene.
Type V Bartter syndrome
Type V Bartter syndrome has been shown to be a digenic disorder resulting from loss-of-function mutations in the genes that encode the chloride channel subunits ClC-Ka and ClC-Kb. [22] The specific genetic defect includes a large deletion in the gene that encodes ClC-Kb (ie, CLCNKB) and a point mutation in the gene that encodes ClC-Ka (CLCNKA).
An etiology of Bartter syndrome that is usually known as autosomal dominant hypocalcemia or autosomal dominant hypoparathyroidism has been described. This type V Bartter syndrome has a gain-of-function mutation in the calcium-sensing receptor (CaSR). The CaSR is expressed in the basolateral membrane of the thick ascending limb of loop of Henle. When this receptor is activated, rate of potassium efflux from ROMK channel is reduced, leading to reduction of Na-K-2Cl cotransporter activity. The lack of luminal positive charge leads to increased level of calcium and magnesium in the urine. The end result is mild renal sodium, chloride, potassium, calcium and magnesium wasting.
This form of Bartter syndrome has additional phenotypic presentation of hypocalcemia and hypomagnesemia. [6, 23]
A summary of currently identified genotype-phenotype correlations in Bartter syndrome is in the table below.
Table 1. Bartter Syndrome Genotype-Phenotype Correlations (Open Table in a new window)
Bartter Syndrome Genotype-Phenotype Correlations |
||
Genetic Type |
Defective Gene |
Clinical Type |
Bartter type I |
NKCC2 |
Neonatal |
Bartter type II |
ROMK |
Neonatal |
Bartter type III |
CLCNKB |
Classic |
Bartter type IV |
BSND |
Neonatal with deafness |
Bartter type IVb |
CLCNKB and CLCNKA |
Neonatal with deafness |
Bartter type V |
CaSR |
Classic |
Epidemiology
International occurrence
Bartter syndrome is rare, and estimates of its occurrence vary from country to country. In the United States, the precise incidence is unknown.
In Costa Rica, the frequency of neonatal Bartter syndrome is approximately 1.2 cases per 100,000 live births but is higher if all preterm births are considered. No evidence of consanguinity was found in the Costa Rican cohort. [24]
In Kuwait, the prevalence of consanguineous marriages or related families in patients with Bartter syndrome is higher than 50%, and prevalence in the general population is 1.7 cases per 100,000 persons. [25]
In Sweden, the frequency has been calculated as 1.2 cases per 1 million persons. Of the 28 patients Rudin reported, 7 came from 3 families; the others were unrelated. [26]
Age-related demographics
Neonatal Bartter syndrome can be suspected before birth or can be diagnosed immediately after birth. In the classic form, symptoms begin in neonates or in infants aged 2 years or younger. Gitelman syndrome is often not diagnosed until adolescence or early adulthood. [27, 28]
Prognosis
Bartter and Gitelman syndromes are autosomal recessive disorders, and neither is curable. The degree of disability depends on the severity of the receptor dysfunction, but the prognosis in many cases is good, with patients able to lead fairly normal lives.
The effects of prostaglandin synthetase inhibition include an increase in the plasma potassium concentration (however, this rarely exceeds 3.5 mEq/L), a decrease in the magnitude of polyuria, and improved general well-being.
With treatment, plasma renin and aldosterone levels normalize. Therapy improves the patient's clinical condition and allows catch-up growth.
Bone age is usually appropriate for chronological age, and pubertal and intellectual development are normal with treatment.
The effectiveness of long-term use of prostaglandin synthetase inhibitors is well established. Some patients may experience a recurrence of hypokalemia, which can be managed by adjusting the indomethacin dose or with potassium supplementation. The disease does not recur in the patient with a transplanted kidney.
A retrospective study of long-term outcome of Bartter syndrome in 54 Korean patients found that the dosage of potassium chloride needed tended to decrease with age, and nephrocalcinosis also decreased. However, on follow-up 8 years after the initial diagnosis, 41% of patients had short stature (height less than 3rd percentile) and 11% had chronic kidney disease, grade 3-5. [29]
Morbidity and mortality
Significant morbidity and mortality occur if Bartter syndrome is untreated. With treatment, the outlook is markedly improved; however, long-term prognosis remains guarded because of the slow progression to chronic renal failure due to interstitial fibrosis.
Sensorineural deafness
Sensorineural deafness associated with Bartter syndrome IV is due to defects in the barttin subunit of the ClC-Ka and CIC-Kb channels. [17, 30]
Nephrocalcinosis
A review of 61 cases of Bartter syndrome reported 29 with nephrocalcinosis, a condition that is often associated with hypercalciuria.
Kidney failure
Kidney failure is fairly uncommon in Bartter syndrome. In a review of 63 patients, 5 developed progressive kidney disease requiring dialysis or transplantation.
In 2 reports of patients who underwent biopsies before developing end-stage renal disease (ESRD), 1 patient had interstitial nephritis, and the other had mesangial and interstitial fibrosis.
One report relates the case of a patient developing reversible acute kidney injury from rhabdomyolysis due to hypokalemia.
Short stature/growth retardation
Nearly all patients with Bartter syndrome have growth retardation. In a review of 66 patients, 62 had growth retardation, often severe (below the fifth percentile for age). Treatment with potassium, indomethacin, and growth hormone (GH) has been effective.
Additional complications
Other complications in Bartter syndrome include the following:
-
Cardiac arrhythmia and sudden death - May result from electrolyte imbalances
-
Failure to thrive and developmental delay - Common in untreated patients
-
Significant decrease in bone mineral density - Has been documented in patients with either the neonatal or classic form
Patient Education
Patients and their parents must understand that no cure exists for the constellation of mutations that causes the various forms of Bartter syndrome. This chronic condition requires taking medications consistently, as prescribed, which is often difficult for children and adolescents. Patients should be aware of potential adverse effects of medical therapy, especially gastrointestinal (GI) irritation and bleeding.
Patients tend to become volume depleted if they are sodium and water restricted. Adequate fluid and electrolyte replacement should be available, especially in hot weather and during exercise. Patients should avoid strenuous exercise because of the danger of dehydration and functional cardiac abnormalities secondary to potassium imbalance.
With regard to diet, patients should be educated about which foods have high potassium content.
Bartter and Gitelman syndromes are autosomal recessive disorders; ie, mutations are required on each allele in the chromosome pair. Offspring carry at least 1 mutated allele. In consanguineous marriages or in marriages between closely related families, genetic counseling may be advisable.
For patient education information, see Growth Hormone Deficiency, Growth Failure in Children, Growth Hormone Deficiency in Children, and Growth Hormone Deficiency FAQs.
-
Normal transport mechanisms in the thick ascending limb of the loop of Henle. Reabsorption of sodium chloride is achieved with the sodium chloride/potassium chloride cotransporter, which is driven by the low intracellular concentrations of sodium, chloride, and potassium. Low concentrations are maintained by the basolateral sodium pump (sodium-potassium adenosine triphosphatase), the basolateral chloride channel (ClC-kb), and the apical potassium channel (ROMK).
-
Type I neonatal Bartter syndrome. Mutations in the sodium chloride/potassium chloride cotransporter gene result in defective reabsorption of sodium, chloride, and potassium.
-
Type II neonatal Bartter syndrome. Mutations in the ROMK gene result in an inability to recycle potassium from the cell back into the tubular lumen, with resultant inhibition of the sodium chloride/potassium chloride cotransporter.
-
Classic Bartter syndrome. Mutations in the ClC-kb chloride channel lead to an inability of chloride to exit the cell, with resultant inhibition of the sodium chloride/potassium chloride cotransporter.
Tables
Bartter Syndrome Genotype-Phenotype Correlations |
||
Genetic Type |
Defective Gene |
Clinical Type |
Bartter type I |
NKCC2 |
Neonatal |
Bartter type II |
ROMK |
Neonatal |
Bartter type III |
CLCNKB |
Classic |
Bartter type IV |
BSND |
Neonatal with deafness |
Bartter type IVb |
CLCNKB and CLCNKA |
Neonatal with deafness |
Bartter type V |
CaSR |
Classic |



