Background
Restrictive lung diseases are characterized by reduced lung volumes, either because of an alteration in lung parenchyma or because of a disease of the pleura, chest wall, or neuromuscular apparatus. Unlike obstructive lung diseases, such as asthma and chronic obstructive pulmonary disease (COPD), which show a normal or increased total lung capacity (TLC), restrictive disease are associated with a decreased TLC. Measures of expiratory airflow are preserved and airway resistance is normal and the forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio is increased. If caused by parenchymal lung disease, restrictive lung disorders are accompanied by reduced gas transfer, which may be marked clinically by desaturation after exercise. [1, 2]
The many disorders that cause reduction or restriction of lung volumes may be divided into two groups based on anatomical structures.
The first is intrinsic lung diseases or diseases of the lung parenchyma. The diseases cause inflammation or scarring of the lung tissue (interstitial lung disease) or result in filling of the air spaces with exudate and debris (pneumonitis). These diseases can be characterized according to etiological factors. They include idiopathic fibrotic diseases, connective-tissue diseases, drug-induced lung disease, environmental exposures (inorganic and organic dusts), and primary diseases of the lungs (including sarcoidosis).
The second is extrinsic disorders or extrapulmonary diseases. The chest wall, pleura, and respiratory muscles are the components of the respiratory pump, and they need to function normally for effective ventilation. Diseases of these structures result in lung restriction, impaired ventilatory function, and respiratory failure (eg, nonmuscular diseases of the chest wall, neuromuscular disorders).
The mnemonic "PAINT" has been used to divide the causes of restrictive lung disease into pleural, alveolar, interstitial, neuromuscular, and thoracic cage abnormalities.
Table 1. Causes of Restrictive Lung Disease (Open Table in a new window)
Causes |
Examples |
Diagnosis |
PFT Findings |
Pleural |
Trapped lung, pleural scarring, large pleural effusions, chronic empyema, asbestosis |
Radiography, CT scanning, pleural manometry, pleural biopsy |
Low RVa, low TLC, low FVC |
Alveolar |
Edema, hemorrhage |
Radiography, CT scanning, physical examination |
Increased DLCOb in hemorrhage (Intrapulmonary hemoglobin absorbs the carbon monoxide, thus increasing the DLCO reading.) |
Interstitial |
Interstitial lung disease including IPFc, NSIPd, COPe |
Radiography, CT scanning, physical examination, echo often shows pulmonary hypertension |
Low RV, low FVC, low TLC, decreased DLCO, poor lung compliance |
Neuromuscular |
Myasthenia gravis, ALSf, myopathy |
Physical examination, EMGsg, serology |
Low RV, low TLC, low NIFh, low MMVi |
Thoracic/extrathoracic |
Obesity, kyphoscoliosis, ascites |
Physical examination |
Low ERVj and FRC in obesity, low VCk, TLC, FRCl in kyphoscoliosis |
aResidual volume. bDiffusion capacity of the lungs for carbon monoxide. cIdiopathic pulmonary fibrosis. dNonspecific interstitial pneumonitis. eCryptogenic organizing pneumonia. fAmyotrophic lateral sclerosis. gElectromyography. hNegative inspiratory force. iMaximal voluntary ventilation. jExpiratory reserve volume. kVital capacity. lFunctional residual capacity. |
|||
Pathophysiology
Air flows to and from the alveoli as lungs inflate and deflate during each respiratory cycle. Lung inflation is accomplished by a contraction of respiratory, diaphragmatic, and external intercostal muscles, whereas deflation is passive at rest. Functional reserve capacity (FRC) is the volume of air in the lungs when the respiratory muscles are fully relaxed and no airflow is present. The volume of FRC is determined by the balance of the inward elastic recoil of the lungs and the outward elastic recoil of the chest wall. Restrictive lung diseases are characterized by a reduction in FRC and other lung volumes because of pathology in the lungs, pleura, or structures of the thoracic cage.
The distensibility of the respiratory system is called compliance. Compliance is the volume change produced by a change in the distending pressure. Lung compliance is independent of the thoracic cage, which is a semirigid container. The compliance of an intact respiratory system is an algebraic sum of the compliances of both of these structures. Therefore, it is influenced by any disease of the lungs, pleura, or chest wall.
In cases of intrinsic lung disease, the physiological effects of diffuse parenchymal disorders reduce all lung volumes by the excessive elastic recoil of the lungs, relative to the outward recoil forces of the chest wall. Expiratory airflow is reduced in proportion to lung volume.
Arterial hypoxemia in disorders of pulmonary parenchyma is primarily caused by ventilation-perfusion mismatching, with further contribution from an intrapulmonary shunt. Decreased diffusion of oxygen rarely contributes to hypoxemia because sufficient time still exists for full equilibration of oxygen or carbon dioxide. However, if transit time is significantly shortened, as with exercise, this can highlight the pathology with significant exercise-induced desaturation.
Etiology
Intrinsic lung diseases
Collagen-vascular diseases, including scleroderma, polymyositis, dermatomyositis, systemic lupus erythematosus, rheumatoid arthritis, and ankylosing spondylitis, are potential causes of restrictive lung disease.
Other causes may include drugs and other treatments (eg, nitrofurantoin, amiodarone, gold, phenytoin, thiazides, hydralazine, bleomycin, bischloroethylnitrosourea [BCNU or carmustine], cyclophosphamide, methotrexate, radiation). Also see Drug-Induced Pulmonary Toxicity.
Causes related to primary or unclassified diseases may include sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis (LAM), pulmonary vasculitis, alveolar proteinosis, eosinophilic pneumonia, and cryptogenic organizing pneumonia (COP).
Inorganic dust exposure (eg, silicosis, asbestosis, talc, pneumoconiosis, berylliosis, hard metal fibrosis, coal worker's pneumoconiosis, chemical worker’s lung) may cause restrictive lung disease.
Organic dust exposure can lead to hypersensitivity pneumonitis (eg, farmer's lung, bird fancier's lung, bagassosis, and mushroom worker's lung, humidifier lung, hot tub pneumonitis).
Idiopathic fibrotic disorders
These may include acute interstitial pneumonia, IPF (usually interstitial pneumonitis), lymphocytic interstitial pneumonitis, desquamative interstitial pneumonitis, and nonspecific interstitial pneumonitis.
Extrinsic disorders
Nonmuscular diseases of the chest wall, in which kyphosis can be idiopathic or secondary, may cause restrictive lung disease. The most common cause of secondary kyphoscoliosis is neuromuscular disease (eg, polio, muscular dystrophy). Fibrothorax, massive pleural effusion, morbid obesity, ankylosing spondylitis, and thoracoplasty are other causes.
Neuromuscular diseases manifest as respiratory muscle weakness and are due to myopathy or myositis, quadriplegia, or phrenic neuropathy from infectious or metabolic causes.
Pleural diseases, including trapped lung or asbestos-related pleural plaques, are an underrecognized, and potentially treatable, cause of restrictive lung disease.
Epidemiology
Frequency
United States
For intrinsic lung diseases, studies cite an overall prevalence of 3-6 cases per 100,000 persons. The prevalence of idiopathic pulmonary fibrosis (IPF) is 27-29 cases per 100,000 persons. [3, 4] The prevalence for adults aged 35-44 years is 2.7 cases per 100,000 persons. Prevalence exceeded 175 cases per 100,000 persons among patients older than 75 years. Exposure to dust, metals, organic solvents, and agricultural employment is associated with increased risk.
In North America, the prevalence of sarcoidosis is 10-40 cases per 100,000 persons. [5]
The incidence of chronic interstitial lung diseases in persons with collagen-vascular diseases is variable, but it is increasing for most diseases. [6]
Kyphoscoliosis is a common extrinsic disorder. It is associated with an incidence of mild deformities amounting to 1 case per 1000 persons, with severe deformity occurring in 1 case per 10,000 persons. [7]
Other nonmuscular and neuromuscular disorders are rare, but their incidence and prevalence are not well known.
According to the US Centers for Disease Control and Prevention (CDC), 35.9% of Americans older than 20 years are obese, and 69% of Americans are at least overweight (body mass index [BMI] 25-30). [8]
International
In Sweden, the prevalence rate for sarcoidosis is 64 cases per 100,000 persons. In Japan, the prevalence rate of sarcoidosis is 10-40 cases per 100,000 persons. [9] The prevalence of sarcoidosis is difficult to determine.
Pediatric data from England have demonstrated the prevalence of idiopathic scoliosis to be 1 case per 200 patients aged 6-14 years. [10]
The worldwide prevalence of fibrotic lung diseases is difficult to determine because studies have not been performed.
Race
Although a familial variant of IPF exists, a genetic predisposition has not yet been elucidated. [11]
The incidence among black Americans is 35.5 cases per 100,000 persons. In contrast, the incidence among white Americans is 10.9 cases per 100,000 persons. [5]
Sex
LAM and lung involvement in tuberous sclerosis occur primarily in premenopausal women, although a handful of cases of LAM have been reported in men. Sporadic LAM has a prevalence of approximately 4.9 cases per 1,000,000 women. Men are more likely to have pneumoconiosis because of occupational exposure, IPF, and collagen-vascular diseases (eg, rheumatoid lung). Worldwide, sarcoidosis is slightly more common in women. [9]
Age
IPF is rare in children. Some intrinsic lung diseases present in patients aged 20-40 years. These include sarcoidosis, collagen-vascular–associated diseases, and pulmonary Langerhans cell histiocytosis (formerly referred to as histiocytosis X). Most patients with IPF are older than 50 years. [12]
Prognosis
The natural history of interstitial lung diseases is variable. It depends on the specific diagnosis and the extent and severity of lung involvement based on high-resolution CT scanning and lung biopsy. [12] IPF is typically a relentless progressive disorder, and patients have a mean survival of 3-6 years after diagnosis. [13] Early recognition of IPF is important to help understand the natural history of the disorder, which may be beneficial for directing patient management and predicting (possibly improving) prognosis. [14]
Pulmonary sarcoidosis has a relatively benign self-limiting course, with spontaneous recovery or stabilization in most cases. [15] However, up to 20% of patients develop pulmonary fibrosis and disability. [16]
Prognosis for collagen-vascular diseases, eosinophilic pneumonia, cryptogenic organizing pneumonia (COP), and drug-induced lung disease is generally favorable with treatment. [17, 18, 19]
Patients with chest wall diseases and neuromuscular disorders develop progressive respiratory failure and often succumb during an intercurrent pulmonary infection. [20]
Mortality/morbidity
The mortality and morbidity from various causes of restrictive lung disease is dependent on the underlying cause of the disease process.
The median survival time for patients with IPF is less than 3 years after diagnosis (noted 3-6 months earlier). Factors that predict poor outcome include older age, male sex, severe dyspnea, history of cigarette smoking, severe loss of lung function, appearance and severity of fibrosis on radiologic studies, lack of response to therapy, and prominent fibroblastic foci on histopathologic evaluation. [13]
See the image below.
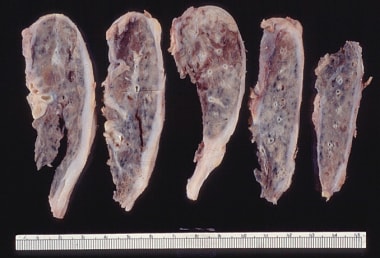 Gross pathology of small and firm lungs due to restrictive lung disease from advanced pulmonary fibrosis.
Gross pathology of small and firm lungs due to restrictive lung disease from advanced pulmonary fibrosis.
-
Approximately half of the patients with idiopathic pulmonary fibrosis develop clubbing. Clubbing is commonly seen in patients with asbestosis.
-
Lung volume is plotted against transpulmonary pressure. Compliance is the change in volume for a given change in pressure. A patient with emphysema has a higher lung compliance compared with a patient with no lung disease, while a patient with restrictive lung disease has a reduction in compliance.
-
Idealized flow volume curves for normal, obstructive, and restrictive lungs.
-
The expiratory flow volume curves of 2 patients are depicted graphically. A is a patient with restrictive lung disease (idiopathic pulmonary fibrosis), low forced vital capacity (FVC), but an increased ratio of forced expiratory volume in 1 second (FEV1) to FVC because of increased elastic recoil. B is a patient with chronic obstructive lung disease whose FEV1/FVC ratio is low but whose lung volumes are increased.
-
Pulmonary function test results from a patient with restrictive lung disease.
-
Gross pathology of small and firm lungs due to restrictive lung disease from advanced pulmonary fibrosis.
-
Intrinsic lung disease may progress to extensive fibrosis, regardless of etiology. This is described as honeycomb lung.
-
End-stage sarcoidosis.
-
Usual interstitial pneumonitis (left).
-
Usual interstitial pneumonitis (right).
-
Histopathology of a case of idiopathic pulmonary fibrosis. Alveolitis with fibroblast proliferation and collagen deposition is present.
-
In usual interstitial pneumonitis or idiopathic pulmonary fibrosis, subpleural and paraseptal inflammation is present, with an appearance of temporal heterogeneity. Patchy scarring of the lung parenchyma and normal, or nearly normal, alveoli interspersed between fibrotic areas are the hallmarks of this disease. Additionally, the lung architecture is completely destroyed.
-
Characteristic features of usual interstitial pneumonitis as described in the image below.
-
Cryptogenic organizing pneumonia (also called proliferative bronchiolitis) is often patchy and peribronchiolar. The proliferation of granulation tissue within small airways and alveolar ducts is excessive and is associated with chronic inflammation of surrounding alveoli.
-
Cryptogenic organizing pneumonia, as described in the image below, showing a close-up view of fibrogranulation tissue in terminal airspaces.
-
Granulomatous lung diseases are marked by granulomas characterized by the accumulation of T lymphocytes, macrophages, and epithelioid cells. These may progress to pulmonary fibrosis. This low-power image shows well-formed granuloma along the airway.
-
Multiple well-formed noncaseating granulomas secondary to sarcoidosis.
-
Sarcoid granulomas.
-
High-power view of sarcoid granuloma shows giant cells.
-
A patient who developed restrictive lung disease had findings of cryptogenic organizing pneumonia on an open lung biopsy specimen.
-
A patient who developed restrictive lung disease had findings of cryptogenic organizing pneumonia on an open lung biopsy specimen. The biopsy sample shows intraluminal buds of granulation tissue.
-
Lymphocytic interstitial pneumonitis, for which the prominent finding is a lymphoid infiltrate that involves both the interstitium and alveolar spaces.
-
Usual interstitial pneumonitis honeycombing.
-
Chest radiograph of a 67-year-old man diagnosed with idiopathic pulmonary fibrosis, based on open lung biopsy findings. Extensive bilateral reticulonodular opacities are seen in both lower lobes.
-
High-resolution CT scan of the same patient in the image below demonstrates peripheral honeycombing and several areas of ground-glass attenuation. Ground-glass opacification may correlate with active alveolitis and a favorable response to therapy.
-
A CT scan image from a 59-year-old woman shows advanced pulmonary fibrosis. Extensive honeycombing and traction bronchiectasis are present.
-
Restrictive lung disease may occur in stage II and stage III sarcoidosis. In this image, mediastinal lymphadenopathy is shown secondary to stage II disease.
-
Sarcoidosis on CT scan shows nodules in midlung zones. These nodules are predominantly along the bronchovascular bundles and in a subpleural location.
-
Restrictive lung disease secondary to sarcoidosis.
-
A chest radiograph of stage III sarcoidosis. This stage refers to pulmonary infiltrates without evidence of mediastinal lymphadenopathy.
-
Chest radiograph from a 39-year-old woman with severe kyphoscoliosis who developed hypercapnic respiratory failure. Spirometry findings showed a severe restrictive lung disease, with a forced expiratory volume in one second of 0.4 L/s and a forced vital capacity of 0.5 L.
-
The flow volume curve of a patient with lung fibrosis.
-
Likely case of idiopathic pulmonary fibrosis, which should be treated with prednisone.
-
Pressure volume curve comparing lungs with emphysema, lungs with restrictive disease, and normal lungs.
Tables
Causes |
Examples |
Diagnosis |
PFT Findings |
Pleural |
Trapped lung, pleural scarring, large pleural effusions, chronic empyema, asbestosis |
Radiography, CT scanning, pleural manometry, pleural biopsy |
Low RVa, low TLC, low FVC |
Alveolar |
Edema, hemorrhage |
Radiography, CT scanning, physical examination |
Increased DLCOb in hemorrhage (Intrapulmonary hemoglobin absorbs the carbon monoxide, thus increasing the DLCO reading.) |
Interstitial |
Interstitial lung disease including IPFc, NSIPd, COPe |
Radiography, CT scanning, physical examination, echo often shows pulmonary hypertension |
Low RV, low FVC, low TLC, decreased DLCO, poor lung compliance |
Neuromuscular |
Myasthenia gravis, ALSf, myopathy |
Physical examination, EMGsg, serology |
Low RV, low TLC, low NIFh, low MMVi |
Thoracic/extrathoracic |
Obesity, kyphoscoliosis, ascites |
Physical examination |
Low ERVj and FRC in obesity, low VCk, TLC, FRCl in kyphoscoliosis |
aResidual volume. bDiffusion capacity of the lungs for carbon monoxide. cIdiopathic pulmonary fibrosis. dNonspecific interstitial pneumonitis. eCryptogenic organizing pneumonia. fAmyotrophic lateral sclerosis. gElectromyography. hNegative inspiratory force. iMaximal voluntary ventilation. jExpiratory reserve volume. kVital capacity. lFunctional residual capacity. |
|||
Features |
AIP |
UIP |
NSIP |
COP |
Pathologic |
||||
Temporal appearance |
Uniform |
Heterogeneous |
Uniform |
Uniform |
Interstitial inflammation |
Scant |
Scant |
Usually prominent |
Variable |
Collagen fibrosis |
No |
Patchy |
Variable, diffuse |
No |
Fibroblast proliferation |
Diffuse, interstitial |
Patchy (fibroblast foci) |
Occasional |
Patchy, airspace |
COP areas |
Rare |
No |
Rare |
-- |
Honeycomb changes |
Rare |
Yes |
Rare |
No |
Hyaline membranes |
Yes, often focal |
No |
No |
No |