Practice Essentials
Relapsing polychondritis (RP) is a severe, episodic, and progressive inflammatory condition involving cartilaginous structures, predominantly those of the ears, nose, and laryngotracheobronchial tree. Other affected structures may include the eyes, cardiovascular system, peripheral joints, skin, middle and inner ear, and central nervous system. [1]
The array of possible presenting symptoms and the episodic nature of relapsing polychondritis may result in a significant delay in diagnosis. In addition, no laboratory findings are specific for relapsing polychondritis. A laboratory evaluation commensurate with the spectrum of reported symptoms is indicated to ascertain the presence of complicating conditions. The mainstay of treatment is systemic corticosteroid therapy.
Background
In 1923, Jaksch-Wartenhorst described a patient who experienced an 18-month course of progressive degeneration of the peripheral joints, external ears, nasal septum, external auditory canals, inner ear, and epiglottis. He termed this condition polychondropathia. [2]
In 1960, Pearson, Kline, and Newcomer reviewed 12 cases and expanded the clinical spectrum of relapsing polychondritis to include nonconcurring inflammation of the auricles, nasal septum, peripheral joints, and larynx, with occasional involvement of the middle and inner ears, the eyes, costal cartilages, spine, trachea, bronchi, and epiglottis. They noted that, after a few episodes of inflammation, the cartilage was replaced by fibrous connective tissue. The term relapsing polychondritis was introduced in that review. [3, 4]
Pathophysiology
The etiology of this rare disease is unknown; however, the pathogenesis is autoimmune. The evidence for an autoimmune etiology includes pathological findings of infiltrating T cells, the presence of antigen-antibody complexes in affected cartilage, cellular and humoral responses against collagen type II and other collagen antigens, and the observation that immunosuppressive regimens most often suppress the disease. [5]
Humoral response
The specificity of autoimmune injury to cartilaginous tissues has led investigators to test the hypothesis that a cartilage-specific autoantibody is central to the pathogenesis of relapsing polychondritis. Various studies find circulating antibodies to cartilage-specific collagen types II, IX, and XI to be present in 30%-70% of patients with relapsing polychondritis. Researchers have found that antibodies to type II collagen are present during acute relapsing polychondritis episodes and that the levels correlate with the severity of the episode. [6]
Treatment with prednisone is associated with a decrease in antibody titers. Antibodies to collagen types I, II, and III are believed to result from cartilage destruction; it has been proposed that antibodies are formed as a primary event in relapsing polychondritis. [6] However, anticollagen type II antibodies are not specific to relapsing polychondritis; they have been identified in other arthritides such as rheumatoid arthritis (RA). The epitope specificity of the antibodies in relapsing polychondritis differs from those in RA, suggesting different mechanisms for formation and pathophysiologic roles.
Autoantibodies to minor cartilage-specific collagens (ie, types IX and XI) have been described. They are more likely to be found in association with antibodies to type II collagen in patients with relapsing polychondritis. Furthermore, levels of antibodies to matrilin 1, an extracellular matrix protein predominantly expressed in tracheal cartilage, were significantly higher in patients with relapsing polychondritis, especially in those with respiratory symptoms, than in patients with Wegener granulomatosis, systemic lupus erythematosus, or RA and in healthy controls. [7]
Most patients with relapsing polychondritis had high titers of antifetal cartilage antibodies during the early acute phase. The antifetal cartilage antibodies were found in 6 of 9 patients and only 4 (1.5%) of 260 patients with RA, exclusively in long-standing disease. [8] A report of relapsing polychondritis in the newborn of a mother with relapsing polychondritis suggests that antibodies crossing the placenta are necessary and sufficient to elicit the entire clinical syndrome.
Using proteomic surveillance to identify ubiquitous cellular proteins in patients with relapsing polychondritis, researchers identified 5 proteins that may be autoantigens. These include (1) tubulin-alpha ubiquitous/6, which, as a family, are main components in microtubules; (2) vimentin, an intermediate filament protein; (3) alpha-enolase; (4) calreticulin, a Ca2+ –binding chaperon indispensable for cardiac development; and (5) colligin-1/2. All but tubulin-alpha have been described as autoantigens in other autoimmune diseases (eg, RA, mixed connective-tissue disease, Behçet disease). Although autoantibodies to tubulin-alpha have been reported in other autoimmune conditions, immunoglobulin G (IgG) antibodies to tubulin-alpha chains are rarely reported and may have diagnostic value in persons with relapsing polychondritis. [9]
Cellular response
Although an inflammatory infiltrate of lymphocytes and neutrophils is the dominant histopathologic feature of relapsing polychondritis, little attention has been paid to the possible role of cellular immune responses in this condition. The association of relapsing polychondritis with HLA-DR4 also suggests an autoimmune pathogenesis. Individuals with HLA-DR4 were found to have a relative risk of 2 for developing relapsing polychondritis. The studies suggest the role of genetic factors in determining risk for developing relapsing polychondritis.
An elegant double-transgenic mouse model provides further evidence that HLA associations are important in the development of relapsing polychondritis. The model demonstrated that more than one HLA class II molecule might be required for expression of susceptibility. The model suggests an important role for cell-mediated immune responses and provides a means for acquiring a detailed understanding of its pathogenesis.
Natural killer T (NKT) cells, lymphocytes discrete from other T, B, and natural killer cells, come in two varieties: CD4+ and CD4-/CD8-. Antigen-presenting cells present antigen to the NKT cells via the major histocompatibility complex–like molecule CD1d. NKT cells are decreased in number and function in several other autoimmune diseases, including multiple sclerosis, RA, systemic lupus erythematosus, systemic sclerosis, and type 1 diabetes mellitus.
Researchers have quantified CD4-/CD8- and CD4+ V-alpha+ V-beta11+ NKT cells and found them decreased in patients with active or quiescent relapsing polychondritis compared with healthy controls. Analysis of the secreted cytokine profile and of binding of alpha-galactosylceramide–loaded CD1d to NKT cells suggests that CD4+ NKT cells play an important role in T1-helper responsiveness in patients with relapsing polychondritis. [10]
Serum levels of 17 cytokines from 22 patients with relapsing polychondritis experiencing a clinical flare were compared with those in age-matched controls. Three of the cytokines, interleukin 8, macrophage inflammatory protein 1-alpha, and monocyte chemoattractant protein-1, were found to be significantly elevated in patients with relapsing polychondritis. All 3 chemokines are proinflammatory and result in accumulation and activation of neutrophils, eosinophils, and monocytes/macrophages. [11]
Additionally, a group of researchers found T cells directed against collagen type II in one patient. A T-cell clone was identified and was found to be specific for a certain region of the collagen type II peptide. This research indicates that a T-cell response to collagen type II may play a role. [12]
Animal models
Mouse and rat models have been helpful in elucidating the autoimmune origin of relapsing polychondritis. Immunization of rats with native bovine type II collagen resulted in bilateral auricular chondritis, with histologic findings similar to the findings of human relapsing polychondritis in 12 of 88 (14%) rats. In addition, 8 of 12 rats developed arthritis. Severe auricular chondritis was accompanied by immunofluorescence positive for IgG and C3 in affected cartilage and by circulating IgG that was reactive against native bovine type II collagen.
Immunization of a different strain of rats with native chick type II collagen was associated with auricular chondritis, in addition to the intended collagen-induced arthritis. Biopsy studies showed that the few auricular lesions contained IgG and C3. Antibodies to native type II collagen were found in the sera of rats that developed auricular chondritis and in rats with collagen-induced arthritis. [13]
Although most data implicate cartilage collagens as the immunogens in relapsing polychondritis, immunization of rats with matrilin 1, a noncollagenous cartilage matrix protein, is associated with development of a clinical syndrome resembling relapsing polychondritis. The syndrome differed significantly from the collagen immunization disease model in that the trachea, nasal cartilages, and kidneys primarily were affected, and the joints and auricles were spared. Matrilin 1 is found in highest levels in the tracheal cartilage and in the nasal septum, likely explaining the observed clinical differences. Matrilin 1 is also found in adult auricular cartilage and costochondral cartilage and is absent in articular cartilage. The presence of both humoral and cellular responses to matrilin 1 has been detected in a patient with significant involvement of the auricular, nasal, and tracheobronchial cartilage and with little arthritis. [14]
The same investigators demonstrated a crucial role for B cells and C5 in the induction of relapsing polychondritis–like symptoms. Additionally, pathogenicity of matrilin 1–specific antibodies in their matrilin 1–induced relapsing polychondritis mouse model was recently recognized. The authors note that further investigation is needed into the role of B cells, complement, and cell-mediated immunity to better understand this complex disease. [14]
Transgenic mice that expressed HLA-DQ6a8b develop spontaneous polychondritis in middle age. This condition is characterized by auricular and nasal chondritis with polyarthritis. As opposed to mice with collagen type II–induced polychondritis, mice with spontaneous polychondritis do not show the overwhelming collagen type II immune response and may serve as a better animal model of relapsing polychondritis. [15]
Other autoimmune disorders
The hypothesis of an autoimmune etiology for relapsing polychondritis is also supported by the high prevalence of other autoimmune disorders found in patients with relapsing polychondritis. McAdam et al reported that 25%-35% of patients with relapsing polychondritis had a concurrent autoimmune disease. [16]
Table. Autoimmune Conditions Reported in Patients With Relapsing Polychondritis (Open Table in a new window)
Disease |
Patients With Condition/Total Patients |
References |
Systemic vasculitis |
3 (5%) of 62 |
Zeuner et al [17] |
11 (10%) of 112 |
Michet et al [18] |
|
8 (12%) of 66 |
Trentham and Le [19] |
|
28 (18%) of 159 |
McAdam et al [16] |
|
50 (13%) of 399 |
Total |
|
Cutaneous leukocytoclastic vasculitis |
2 (33%) of 6 |
Priori et al [20] |
6 (5%) of 112 |
Michet et al [18] |
|
8 (7%) of 118 |
Total |
|
Thyroid disease |
8 (5%) of 159 |
McAdam et al [16] |
10 (15%) of 66 |
Trentham and Le [19] |
|
2 (33%) of 6 |
Priori et al [20] |
|
4 (4%) of 112 |
Michet et al [18] |
|
2 (3%) of 62 |
Zeuner et al [17] |
|
26 (6%) of 405 |
Total |
|
Rheumatoid arthritis* |
8 (5%) of 159 |
McAdam et al [16] |
3 (2%) of 180 |
Piette et al [21] |
|
8 (7%) of 112 |
Michet et al [18] |
|
7 (11%) of 62 |
Zeuner et al [17] |
|
26 (5%) of 513 |
Total |
|
Systemic lupus erythematosus† |
2 (1%) of 159 |
McAdam et al [16] |
9 (5%) of 180 |
Piette et al [21] |
|
1 (17%) of 6 |
Priori et al [20] |
|
6 (5%) of 112 |
Michet et al [18] |
|
3 (5%) of 62 |
Zeuner et al [17] |
|
21 (4%) of 519 |
Total |
|
Sjögren syndrome (possible) |
5 (3%) of 159 |
McAdam et al [16] |
5 (5%) of 111 |
Piette et al [21] |
|
10 (4%) of 270 |
Total |
|
3 (2%) of 159 |
McAdam et al [16] |
|
2 (3%) of 62 |
Zeuner et al [17] |
|
5 (2%) of 221 |
Total |
|
2 (1%) of 180 |
Piette et al [21] |
|
1 (2%) 62 |
Zeuner et al [17] |
|
1 (100%) of 1 |
Haigh et al [22] |
|
4 (2%) of 243 |
Total |
|
Mixed connective-tissue disease |
5 (3%) of 180 |
Piette et al [21] |
2 (2%) of 112 |
Michet et al [18] |
|
7 (2%) of 292 |
Total |
|
3 (2%) of 180 |
Piette et al [21] |
|
Mesenteric panniculitis |
3 (2%) of 180 |
Piette et al [21] |
Spondyloarthropathy |
2 (1%) of 180 |
Piette et al [21] |
3 (3%) of 112 |
Michet et al [18] |
|
2 (3%) of 62 |
Zeuner et al [17] |
|
7 (2%) of 354 |
Total |
|
Diabetes mellitus |
1 (2%) of 62 |
Zeuner et al [17] |
3 (2%) of 159 |
McAdam et al [16] |
|
4 (2%) of 221 |
Total |
|
2 (1%) of 159 |
McAdam et al [16] |
|
1 (< 1%) of 112 |
Michet et al [18] |
|
3 (1%) of 271 |
Total |
|
2 (1%) of 159 |
McAdam et al [16] |
|
2 (1%) of 159 |
McAdam et al [16] |
|
Glomerulonephritis |
2 (1%) of 159 |
McAdam et al [16] |
Dysgammaglobulinemia |
2 (1%)of 159 |
McAdam et al [16] |
1 (1%) of 159 |
McAdam et al [16] |
|
Behçet disease* |
1 (< 1%) of 112 |
Michet et al [18] |
Psoriasis |
2 (1%) of 180 |
Piette et al [21] |
2 (1%) of 180 |
Piette et al [21] |
|
1 (< 1%) of 112 |
Michet et al [18] |
|
*Individual patients may carry more than one autoimmune diagnosis. †Reported as 13 (20%) of 66 prevalence by Trentham and Le without division by disease |
||
In addition, several reports have linked relapsing polychondritis with internal malignancy. It is thought to be paraneoplastic in these cases. The underlying malignancy is most often hematological in nature, but solid tumors have also been described. [23]
In 2020, Beck and colleagues described a genetic disorder they termed VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome. VEXAS syndrome results from somatic mutations in thegene that codes for UBA1, the major E1 enzyme that initiates ubiquitylation. In late adulthood, these patients develop an often fatal, treatment-refractory inflammatory syndrome (eg, relapsing polychondritis), a hematologic condition (myelodysplastic syndrome or multiple myeloma), or both. This could explain the co-occurrence of relapsing polychondritis with myelodysplasia. [24]
Gut microbiome alterations
Shimizu et al propose that the pathogenesis of relapsing polychondritis may involve alteration of gut microbiota. Their study found that the gut microbiome in patients with relapsing polychondritis contains higher numbers of microbes that produce propionate, which is a short-chain fatty acid that may affect interleukin-10 (IL-10)–producing regulatory T (Treg) cell differentiation in gut-associated lymphoid tissues. [25]
These authors suggest that in relapsing polychondritis, continuous stimulation of intestinal T cells by excessive propionate leads to the spontaneous production of IL-10 and a subsequent refractory period of T cells. In turn, hyporesponsiveness of Treg cells upon activation may associate with production by PBMC and subsequent chondritis. [25]
our findings suggested that propionate-producing gut microbes became predominant, leading to defective Treg cell function upon activation in patients with RP. Decreased production of IL10 by Treg cells and increased production of the inflammatory cytokine tumor necrosis factor–α TNFα by PBMC may lead to chondritis in patients with relapsing polychondritis. [25]
Etiology
The cause of relapsing polychondritis is not known. Familial clustering has not been observed. A genotyping study by Terao et al found that HLA-DRB1*16:02, HLA-DQB1*05:02 and HLA-B*67:01, in linkage disequilibrium with each other, are associated with susceptibility to relapsing polychondritis. In Germans, susceptibility for developing relapsing polychondritis has been found to be increased slightly in carriers of the HLA-DR4 haplotype. [26]
Three intriguing case reports suggest that hormonal influences may be important in relapsing polychondritis. Two men have developed relapsing polychondritis after receiving injections of luteinizing hormone–releasing hormone, and a woman with arthritis mutilans had a sudden exacerbation of her condition and new onset of atrophy of the auricular cartilage, nasal septum, weight loss, and deafness after receiving an injection of chorionic gonadotropin. [27]
Epidemiology
In clinical reports and reviews, relapsing polychondritis is reported to be a rare disease. The annual incidence in Rochester, Minnesota, was noted to be 3.5 cases per million population. [28]
Relapsing polychondritis is most common in whites. Although the disorder has been found in persons of all races, few data are available for nonwhite persons.
Reviews from the 1970s and 1980s found that relapsing polychondritis has no sexual predilection. However, reviews in 1998 and 2002 suggested a slight female predominance. [29, 19] Saddle-nose deformity and subglottic stricture are more common in females.
Relapsing polychondritis may occur at any age, but onset is usually in the fourth or fifth decade of life. No relationship exists between age of onset and sex. [1]
Prognosis
In earlier studies, the 5-year survival rate associated with relapsing polychondritis was reported to be 66%-74% (45% if relapsing polychondritis occurs with systemic vasculitis), with a 10-year survival rate of 55%. More recently, Trentham and Le found a survival rate of 94% at 8 years. [19] However, those data may represent relapsing polychondritis in patients with less severe disease than patients studied in earlier reports.
A study analyzing mortality rates of relapsing polychondritis based on organ involvement at disease onset reported that the 5-year mortality was higher in patients with respiratory onset (15%) and lowest in patients with auricular onset (5.9%). The mortality rate in patients with CNS, ocular, and inner ear involvment, among others, was also 15%. [30]
The most common causes of relapsing polychondritis–related death include infection secondary to corticosteroid treatment or respiratory compromise (10-50% of deaths result from airway complications), systemic vasculitis, and malignancy unrelated to relapsing polychondritis.
Although the life expectancy in all patients with relapsing polychondritis is decreased compared with age- and sex-matched healthy individuals, patients with renal involvement have a significantly lower age-adjusted life expectancy. In those with renal disease, uremia is the third most frequent cause of death.
Dion et al identified three factors associated with death in patients with relapsing polychondritis, as follows [31] :
-
Male sex
-
Cardiac abnormalities
-
Concomitant myelodysplastic syndrome or other hematologic malignancy
Complications of relapsing polychondritis such as saddle-nose deformity (see the image below), systemic vasculitis, laryngotracheobronchial stricture, arthritis, and anemia in patients younger than 51 years portend a poorer prognosis than in age-matched patients with relapsing polychondritis without complications. In patients older than 51 years, only anemia is associated with a poorer prognosis. Renal involvement is a poor prognostic factor at all ages.
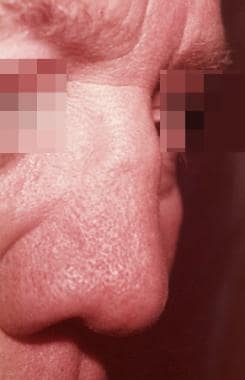 Saddle-nose deformity. Courtesy of the University of Washington, Division of Dermatology.
Saddle-nose deformity. Courtesy of the University of Washington, Division of Dermatology.
Complications of relapsing polychondritis include the following:
-
Vertigo
-
Tinnitus
-
Voice hoarseness
-
Joint deformity
-
Epiglottitis
-
Scleritis
-
Conjunctivitis
-
Iritis
-
Need for permanent tracheotomy (severe cases)
-
Severe pulmonary infection
-
Blindness
-
Frail chest wall
-
Respiratory failure
-
Aortic dissection
-
Glomerulonephritis-associated renal failure
-
Auricular edema and erythema sparing the lobule. Courtesy of Gregory J. Raugi, MD, PhD.
-
Severe auricular edema and inflammation. Courtesy of the University of Washington, Division of Dermatology.
-
Forward listing ear. Courtesy of the University of Washington, Division of Dermatology.
-
Floppy ear. Courtesy of the University of Washington, Division of Dermatology.
-
Bilateral inflammation and structural collapse of the auricles in a patient found to have aortic dissection. Courtesy of the University of Washington, Division of Dermatology.
-
Same patient as in Image 5 after 4-6 weeks of steroid treatment. Note resolution of auricular inflammation with nodularity and forward listing of the ears. Courtesy of the University of Washington, Division of Dermatology.
-
Close-up view of same patient as in Image 6. Forward flopping of ear with nodularity after steroid treatment. Courtesy of the University of Washington, Division of Dermatology.
-
Unilateral episcleritis. Courtesy of Gregory J. Raugi, MD, PhD.
-
Saddle-nose deformity. Courtesy of the University of Washington, Division of Dermatology.
-
Tracheal stenosis on chest x-ray film. Courtesy of Julie E. Takasugi, MD.
Tables
Disease |
Patients With Condition/Total Patients |
References |
Systemic vasculitis |
3 (5%) of 62 |
Zeuner et al [17] |
11 (10%) of 112 |
Michet et al [18] |
|
8 (12%) of 66 |
Trentham and Le [19] |
|
28 (18%) of 159 |
McAdam et al [16] |
|
50 (13%) of 399 |
Total |
|
Cutaneous leukocytoclastic vasculitis |
2 (33%) of 6 |
Priori et al [20] |
6 (5%) of 112 |
Michet et al [18] |
|
8 (7%) of 118 |
Total |
|
Thyroid disease |
8 (5%) of 159 |
McAdam et al [16] |
10 (15%) of 66 |
Trentham and Le [19] |
|
2 (33%) of 6 |
Priori et al [20] |
|
4 (4%) of 112 |
Michet et al [18] |
|
2 (3%) of 62 |
Zeuner et al [17] |
|
26 (6%) of 405 |
Total |
|
Rheumatoid arthritis* |
8 (5%) of 159 |
McAdam et al [16] |
3 (2%) of 180 |
Piette et al [21] |
|
8 (7%) of 112 |
Michet et al [18] |
|
7 (11%) of 62 |
Zeuner et al [17] |
|
26 (5%) of 513 |
Total |
|
Systemic lupus erythematosus† |
2 (1%) of 159 |
McAdam et al [16] |
9 (5%) of 180 |
Piette et al [21] |
|
1 (17%) of 6 |
Priori et al [20] |
|
6 (5%) of 112 |
Michet et al [18] |
|
3 (5%) of 62 |
Zeuner et al [17] |
|
21 (4%) of 519 |
Total |
|
Sjögren syndrome (possible) |
5 (3%) of 159 |
McAdam et al [16] |
5 (5%) of 111 |
Piette et al [21] |
|
10 (4%) of 270 |
Total |
|
3 (2%) of 159 |
McAdam et al [16] |
|
2 (3%) of 62 |
Zeuner et al [17] |
|
5 (2%) of 221 |
Total |
|
2 (1%) of 180 |
Piette et al [21] |
|
1 (2%) 62 |
Zeuner et al [17] |
|
1 (100%) of 1 |
Haigh et al [22] |
|
4 (2%) of 243 |
Total |
|
Mixed connective-tissue disease |
5 (3%) of 180 |
Piette et al [21] |
2 (2%) of 112 |
Michet et al [18] |
|
7 (2%) of 292 |
Total |
|
3 (2%) of 180 |
Piette et al [21] |
|
Mesenteric panniculitis |
3 (2%) of 180 |
Piette et al [21] |
Spondyloarthropathy |
2 (1%) of 180 |
Piette et al [21] |
3 (3%) of 112 |
Michet et al [18] |
|
2 (3%) of 62 |
Zeuner et al [17] |
|
7 (2%) of 354 |
Total |
|
Diabetes mellitus |
1 (2%) of 62 |
Zeuner et al [17] |
3 (2%) of 159 |
McAdam et al [16] |
|
4 (2%) of 221 |
Total |
|
2 (1%) of 159 |
McAdam et al [16] |
|
1 (< 1%) of 112 |
Michet et al [18] |
|
3 (1%) of 271 |
Total |
|
2 (1%) of 159 |
McAdam et al [16] |
|
2 (1%) of 159 |
McAdam et al [16] |
|
Glomerulonephritis |
2 (1%) of 159 |
McAdam et al [16] |
Dysgammaglobulinemia |
2 (1%)of 159 |
McAdam et al [16] |
1 (1%) of 159 |
McAdam et al [16] |
|
Behçet disease* |
1 (< 1%) of 112 |
Michet et al [18] |
Psoriasis |
2 (1%) of 180 |
Piette et al [21] |
2 (1%) of 180 |
Piette et al [21] |
|
1 (< 1%) of 112 |
Michet et al [18] |
|
*Individual patients may carry more than one autoimmune diagnosis. †Reported as 13 (20%) of 66 prevalence by Trentham and Le without division by disease |
||







