Practice Essentials
Long QT syndrome (LQTS) is a congenital disorder characterized by a prolongation of the QT interval on electrocardiograms (ECGs) and a propensity to ventricular tachyarrhythmias, which may lead to syncope, cardiac arrest, or sudden death. See the image below.
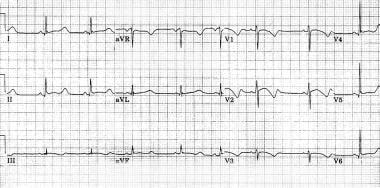 Marked prolongation of QT interval in a 15-year-old male adolescent with long QT syndrome (LQTS) (R-R = 1.00 s, QT interval = 0.56 s, QT interval corrected for heart rate [QTc] = 0.56 s). Abnormal morphology of repolarization can be observed in almost every lead (ie, peaked T waves, bowing ST segment). Bradycardia is a common feature in patients with LQTS.
Marked prolongation of QT interval in a 15-year-old male adolescent with long QT syndrome (LQTS) (R-R = 1.00 s, QT interval = 0.56 s, QT interval corrected for heart rate [QTc] = 0.56 s). Abnormal morphology of repolarization can be observed in almost every lead (ie, peaked T waves, bowing ST segment). Bradycardia is a common feature in patients with LQTS.
See 7 Can't-Miss Life-Threatening ECG Findings, a Critical Images slideshow, to help recognize the conditions shown in various tracings.
Signs and symptoms
LQTS is usually diagnosed after a person has a cardiac event (eg, syncope, cardiac arrest). In some situations, this condition is diagnosed after a family member suddenly dies. In some individuals, the diagnosis is made when an ECG shows QT prolongation.
A history of cardiac events is the most typical clinical presentation in patients with LQTS.
See Presentation for more detail.
Diagnosis
Findings on physical examination usually do not indicate a diagnosis of LQTS, although some patients may present with excessive bradycardia for their age, and some patients may have hearing loss (congenital deafness), indicating the possibility of Jervell and Lange-Nielsen syndrome. Skeletal abnormalities, such as short stature and scoliosis are seen in the LQT7 type (Andersen syndrome), and congenital heart diseases, cognitive and behavioral problems, musculoskeletal diseases, and immune dysfunction may be seen in those with LQT8 type (Timothy syndrome).
Testing
Diagnostic studies in patients with suspected LQTS include the following:
-
Serum potassium and magnesium levels
-
Thyroid function tests
-
Pharmacologic provocation with epinephrine or isoproterenol in patients with a borderline presentation
-
Electrocardiography of the patient and family members
-
Genetic testing of the patient and family members
An increased corrected QT (QTc) interval in response to standing up (“response to standing” test), which is associated with increased sympathetic tone, can provide more diagnostic information in patients with LQTS. [1] This increase in QTc in response to standing may persist in these patients even after their heart rate returns to normal. [2]
See Workup for more detail.
Management
No treatment addresses the cause of LQTS. Antiadrenergic therapeutic measures (eg, use of beta-blockers, left cervicothoracic stellectomy) and device therapy (eg, use of pacemakers, implantable cardioverter-defibrillators) aim to decrease the risk and lethality of cardiac events.
Pharmacotherapy
Beta-adrenergic blocking agents are the drugs of choice to treat LQTS and include the following medications:
-
Nadolol
-
Propranolol
-
Metoprolol
-
Atenolol
Nadolol, however, is the preferred beta-blocker to be used at a dose of 1-1.5 mg/kg/day (once a day for patients older than 12 years; divided twice a day for younger patients). [3]
Surgical option
Surgical intervention in patients with LQTS may include the following procedures:
-
Implantation of cardioverter-defibrillators
-
Placement of a pacemaker
-
Left cervicothoracic stellectomy
Nonpharmacotherapy
Patients with LQTS should avoid participation in competitive sports, strenuous exercise, and stress-related emotions.
These individuals should also avoid the following agents:
-
Anesthetics or asthma medication (eg, epinephrine)
-
Antihistamines (eg, diphenhydramine; terfenadine and astemizole [both recalled from the US market])
-
Antibiotics (eg, erythromycin, trimethoprim and sulfamethoxazole, pentamidine)
-
Cardiac medications (eg, quinidine, procainamide, disopyramide, sotalol, probucol, bepridil, dofetilide, ibutilide)
-
Gastrointestinal medications (eg, cisapride)
-
Antifungal medications (eg, ketoconazole, fluconazole, itraconazole)
-
Psychotropic medications (eg, tricyclic antidepressants, phenothiazine derivatives, butyrophenones, benzisoxazole, diphenylbutylpiperidine)
-
Potassium-losing medications (eg, indapamide, other diuretics; medications for vomiting/diarrhea)
See Treatment and Medication for more detail.
Background
Long QT syndrome (LQTS) is a congenital disorder characterized by a prolongation of the QT interval on electrocardiograms (ECGs) and a propensity to ventricular tachyarrhythmias, which may lead to syncope, cardiac arrest, or sudden death. (See Etiology, Prognosis, Presentation, and Workup.)
The QT interval on the ECG, measured from the beginning of the QRS complex to the end of the T wave, represents the duration of activation and recovery of the ventricular myocardium. A QT interval corrected for heart rate (QTc) that is longer than 0.44 seconds is generally considered to be abnormal, although a normal QTc can be more prolonged in females (up to 0.46sec). The Bazett formula is the formula most commonly used to calculate the QTc, as follows: QTc = QT/square root of the R-R interval (in seconds). (See Workup.)
To measure the QT interval accurately, the relationship of QT to the R-R interval should be reproducible. This issue is especially important when the heart rate is lower than 50 beats per minute (bpm) or over 120 bpm, and when athletes or children have marked beat-to-beat variability of the R-R interval. In such cases, long recordings and several measurements are required. The longest QT interval is usually observed in the right precordial leads. When marked variation is present in the R-R interval (atrial fibrillation, ectopy), correction of the QT interval is difficult to define precisely. (See Workup.)
Etiopathophysiology
The QT interval represents the duration of activation and recovery of the ventricular myocardium. Prolonged recovery from electrical excitation increases the likelihood of dispersing refractoriness, when some parts of myocardium might be refractory to subsequent depolarization.
From a physiologic standpoint, dispersion occurs with repolarization between three layers of the heart, and the repolarization phase tends to be prolonged in the mid myocardium. This is why the T wave is normally wide and the interval from Tpeak to Tend (Tp-e) represents the transmural dispersion of repolarization (TDR). In long QT syndrome (LQTS), TDR increases and creates a functional substrate for transmural reentry.
Hypokalemia, hypocalcemia, and use of loop diuretics are risk factors for QTc interval prolongation. [4]
LQTS has been recognized as mainly Romano-Ward syndrome (ie, familial occurrence with autosomal dominant inheritance, QT prolongation, and ventricular tachyarrhythmias) or as Jervell and Lang-Nielsen (JLN) syndrome (ie, familial occurrence with autosomal recessive inheritance, congenital deafness, QT prolongation, and ventricular arrhythmias). Two other syndromes are described, namely, Andersen syndrome and Timothy syndrome, although there is some debate on whether they should be included in LQTS.
Torsade de pointes
In LQTS, QT prolongation can lead to polymorphic ventricular tachycardia, or torsade de pointes, which itself may lead to ventricular fibrillation and sudden cardiac death. Torsade de pointes is widely thought to be triggered by reactivation of calcium channels, reactivation of a delayed sodium current, or a decreased outward potassium current that results in early afterdepolarization (EAD), in a condition with enhanced TDR usually associated with a prolonged QT interval. [5] TDR serves as a functional reentry substrate to maintain torsade de pointes.
TDR not only provides a substrate for reentry but also increases the likelihood of EAD, the triggering event for torsade de pointes, by prolonging the time window for calcium channels to remain open. Any additional condition that accelerates the reactivation of calcium channels (eg, increased sympathetic tone) increases the risk of EAD.
Genetics
LQTS is known to be caused by mutations of the genes for cardiac potassium, sodium, or calcium ion channels; at least 10 genes have been identified. Based on this genetic background, 6 types of Romano-Ward syndrome, 1 type of Andersen syndrome and 1 type of Timothy syndrome, and 2 types of JLN syndrome are characterized (see Table 1, below).
LQTS results from mutations of genes encoding for cardiac ion channel proteins, which cause abnormal ion channel kinetics. Shortened opening of the potassium channel in LQT1, LQT2, LQT5, LQT6, JLN1, and JLN2 and delayed closing of a sodium channel in LQT3 overcharges a myocardial cell with positive ions. At least 10 genes have been identified in LQTS.
Table 1. Genetic Background of Inherited Forms of LQTS (Romano-Ward Syndrome: LQT1-6, Anderson Syndrome: LQT7, Timothy Syndrome: LQT8, and Jervell and Lang-Nielsen Syndrome: JLN1-2) (Open Table in a new window)
Type of LQTS |
Chromosomal Locus |
Mutated Gene |
Ion Current Affected |
LQT1 |
11p15.5 |
KVLQT1 or KCNQ1 (heterozygotes) |
Potassium (IKs) |
LQT2 |
7q35-36 |
HERG, KCNH2 |
Potassium (IKr) |
LQT3 |
3p21-24 |
SCN5A |
Sodium (INa) |
LQT4 |
4q25-27 |
ANK2, ANKB |
Sodium, potassium and calcium |
LQT5 |
21q22.1-22.2 |
KCNE1 (heterozygotes) |
Potassium (IKs) |
LQT6 |
21q22.1-22.2 |
MiRP1, KNCE2 |
Potassium (IKr) |
LQT7 (Anderson syndrome) |
17q23.1-q24.2 |
KCNJ2 |
Potassium (IK1) |
LQT8 (Timothy syndrome) |
12q13.3 |
CACNA1C |
Calcium (ICa-Lalpha) |
LQT9 |
3p25.3 |
CAV3 |
Sodium (INa) |
LQT10 |
11q23.3 |
SCN4B |
Sodium (INa) |
LQT11 |
7q21-q22 |
AKAP9 |
Potassium (IKs) |
LQT12 |
|
SNTAI |
Sodium (INa) |
JLN1 |
11p15.5 |
KVLQT1 or KCNQ1 (homozygotes) |
Potassium (IKs) |
JLN2 |
21q22.1-22.2 |
KCNE1 (homozygotes) |
Potassium (IKs) |
LQT1, LQT2, and LQT3 account for most cases of LQTS, with estimated prevalences of 45%, 45%, and 7%, respectively. In LQTS, QT prolongation is due to an overload of myocardial cells with positively charged ions during ventricular repolarization. In LQT1, LQT2, LQT5, LQT6, and LQT7, potassium ion channels are blocked, they open with a delay, or they are open for a shorter period than they are in normally functioning channels. These changes decrease the potassium outward current and prolong repolarization.
LQT1
The LQT1 gene (KVLQT1 or KCNQ1) encodes for part of the IKs slowly deactivating, delayed rectifier potassium channel. [6] More than 170 mutations (most missense) of this gene have been reported. Their net effect is a decreased outward potassium current. Therefore, the channels remain open longer than usual, with a delay in ventricular repolarization and with QT prolongation.
LQT2
The LQT2 gene (HERG or KCNH2) encodes for part of IKr rapidly activating, rapidly deactivating, delayed rectifier potassium channel. [7, 8] Mutations in this gene cause rapid closure of the potassium channels and decrease the normal rise in IKr. They also result in delayed ventricular repolarization and QT prolongation. About 200 mutations in this gene have been detected.
Japanese researchers have identified two novel KCNH2 missense mutations, G785D and T826I, that disrupt the intracellular transport of KV11.1 to the plasma membrane; low-temperature incubation appears to restore plasma membrane expression of Kv11.1-T826I but not G785D. [9]
More than 30% of identified LQT2 mutations consist of nonsense or frameshift mutations that introduce premature termination codons (PTCs), which lead to the degradation of mutant mRNA by nonsense-mediated mRNA decay, an RNA surveillance mechanism that selectively eliminates the mRNA transcripts that contain PTCs. [8]
LQT3
In LQT3, caused by mutations of the SCN5A gene for the sodium channel, a gain-of-function mutation causes persistent inward sodium current in the plateau phase, which contributes to prolonged repolarization. [10] Some loss-of-function mutations in the same gene may lead to different presentations, including Brugada syndrome. More than 50 mutations have been identified in this gene.
Cardiac events are less frequent in congenital LQT3 than in LQT1 and LQT2, but they are more likely to be lethal (20% mortality with cardiac events in families with LQT3 mutations; 4% in those with an LQT1 or LQT2 mutation). [10]
In some patients, caveolae proteins have been recognized as responsible for the increased sodium current in LQTS3. [11] Caveolae are small (50-100 nm) microdomains that exist on the membrane of a variety of cells, including cardiac myocytes and fibroblasts. Some ion channels, and in particular the SCN5A-encoded voltage-gated sodium channels, are mainly co-localized with caveolae on the membrane. Thus, absence or abnormal formation of caveolae may have some effects on the availability of sodium channels. For example, Vatta and colleagues demonstrated that mutations in caveolin-3 protein exist in LQTS3 and that they can cause an increase in late sodium current. [11]
Nevertheless, caveolae are present in the membrane of many other cell types and are also involved in many cellular activities, thus, their impairment is expected to be associated with multisystemic diseases. For example, Rajab and colleagues reported genetic mutations resulting in defective caveolae in families with congenital generalized lipodystrophy who have several systemic manifestations, such as hypertrophic pyloric stenosis, impaired bone formations, ventricular arrhythmia, and sudden cardiac death. [12] The fact that mutations in proteins associated with ion channels may result in a change in the availability of channels on the membrane, and therefore a significant change in total current, has added another window for investigating the genetic abnormalities resulting in LQTS.
LQT4 gene
The LQT4 gene (ANK2 or ANKB) encodes for ankyrin-B. Ankyrins are adapter proteins that bind to several ion channel proteins, such as the anion exchanger (chloride-bicarbonate exchanger), sodium-potassium adenosine triphosphatase (ATPase), the voltage-sensitive sodium channel (INa), the sodium-calcium exchanger (NCX, or INa-Ca), and calcium-release channels (including those mediated by the receptors for inositol triphosphate [IP3] or ryanodine).
Mutations in this gene interfere with several of these ion channels. The end result is increased intracellular concentration of calcium and, sometimes, fatal arrhythmia. Five mutations of this gene are reported. LQT4 is interesting, because it provides an example of how mutations in proteins other than ion channels can be involved in the pathogenesis of LQTS.
LQT5, 6, 7, 8, 9, 10
The LQT5 gene encodes for the IKs potassium channel. Similar to LQT1, LQT5 is involved in a decreased outward current of potassium and in QT prolongation.
LQT6 involves mutations in the gene MiRP1, or KCNE2, which encodes for the potassium channel beta subunit MinK-related protein 1 (MiRP1). KCNE2 encodes for the beta subunits of IKr potassium channels.
The LQT7 gene (KCNJ2) encodes for potassium channel 2 protein, which plays an important role in inward repolarizing current (IKi), especially in phase 3 of the action potential. In this subtype, QT prolongation is less prominent than in other types, and the QT interval is sometimes in the normal range. Because potassium channel 2 protein is expressed in cardiac and skeletal muscle, Andersen syndrome is associated with skeletal abnormalities, such as short stature and scoliosis.
Mutations in the LQT8 gene (CACNA1C) cause loss of L-type calcium current. So far, a limited number of cases of Timothy syndrome have been reported. They have been associated with abnormalities such as congenital heart disease, cognitive and behavioral problems, musculoskeletal diseases, and immune dysfunction.
The LQT9 gene encodes for caveolin 3, a caveolae plasma membrane component protein involved in scaffolding proteins. The voltage-gated sodium channel (NaV b3) is associated with this protein. Functional studies have demonstrated that CAV3 mutations are associated with persistent late sodium current, and they have been reported in cases of sudden infant death syndrome (SIDS). [13] LQT9 and LQT4 serve as examples of LQTS with nonchannel mutations.
A novel mutation in the LQT10 gene encoding the protein NaV b4, a subunit of the voltage-gated sodium channel of the heart, NaV 1.5 (gene SCN5), results in a positive shift in the inactivation of the sodium current. To date, only a single mutation in one patient has been described. [14]
Alpha-1-syntrophic gene mutation
The newest genetic missense mutation associated with LQTS has been described in the alpha-1-syntrophin gene and results in gain of function of the sodium channel similar to that observed in LQT3. [15]
Stimuli
In patients with LQTS, a variety of adrenergic stimuli, including exercise, emotion, loud noise, and swimming, may precipitate an arrhythmic response. However, arrhythmia may occur without such preceding conditions.
Drug-induced QT prolongation
Secondary (drug-induced) QT prolongation may also increase the risk of ventricular tachyarrhythmias (eg, torsade de pointes) and sudden cardiac death. The ionic mechanism is similar to that observed in congenital LQTS (ie, mainly intrinsic blockade of cardiac potassium efflux).
In addition to the medications that potentially can prolong the QT interval, several other factors play a role in this phenomenon. Important risk factors for drug-induced QT prolongation include the following:
-
Female sex [4]
-
Electrolyte disturbances (hypokalemia and hypomagnesemia)
-
Hypothermia
-
Abnormal thyroid function
-
Structural heart disease
-
Bradycardia
Drug-induced QT prolongation may also have a genetic background, consisting of the predisposition of an ion channel to abnormal kinetics caused by gene mutation or polymorphism. However, data are insufficient to claim that all patients with drug-induced QT prolongation have a genetic LQTS-related mechanism. The Arizona Center for Education and Research on Therapeutics (AZCERT) provides lists of drugs that prolong the QT interval and/or induce torsade de pointes ventricular arrhythmia.
Epidemiology
United States data
Long QT syndrome (LQTS) remains an underdiagnosed disorder, especially because some individuals may LQTS gene mutation carriers who have a normal QTc duration. [16]
The prevalence of LQTS is difficult to estimate. However, LQTS may be expected to occur in the range of 1 in 2,000 to 1 in 5,000 individuals. [16, 17]
International data
The occurrence of long QT syndrome internationally is similar to that in the United States.
Sex-related demographics
Newly diagnosed cases of LQTS are more prevalent in female patients (60-70% of cases) than in male patients. The female predominance may be related to the relatively prolonged QTc (as determined by using the Bazett formula) in women compared to men and to a relatively higher mortality rate in young men.
In women, pregnancy is not associated with an increased incidence of cardiac events, whereas the postpartum period is associated with a substantially increased risk of cardiac events, especially in the subset of patients with LQT2. Cardiac events have been highly correlated with menses.
In addition, a significantly higher risk of cardiac events (a 3- to 8-fold increase, mainly in the form of recurrent episodes of syncope) has been reported in women with LQT2 syndrome during and after the onset of menopause, compared with the reproductive years. [18]
Age-related demographics
Patients with LQTS usually present with cardiac events (eg, syncope, aborted cardiac arrest, sudden death) in childhood, adolescence, or early adulthood. However, LQTS has been identified in adults as late as in the fifth decade of life. The risk of death from LQTS is higher in boys than in girls younger than 10 years; the risk is similar in male and female patients thereafter.
Prognosis
The prognosis is good overall for patients with long QT syndrome (LQTS) treated with beta-blockers (and other therapeutic measures, if needed). Fortunately, episodes of torsade de pointes are usually self-terminating in patients with LQTS; only about 4-5% of cardiac events are fatal.
Patients at high risk (ie, those with aborted cardiac arrest or recurrent cardiac events despite beta-blocker therapy) have a markedly increased risk of sudden death. Treat these patients with an implantable cardioverter-defibrillator (ICDs); their prognosis after implantation of an ICD is good.
Morbidity and mortality
Mortality, morbidity, and responses to pharmacologic treatment differ in the various types of LQTS. This issue is under investigation.
LQTS may result in syncope and lead to sudden cardiac death, which usually occurs in otherwise healthy young individuals. LQTS is thought to cause about 4,000 deaths in the United States each year. The cumulative mortality rate reaches approximately 6% by the age of 40 years.
Although sudden death usually occurs in symptomatic patients, it can also materialize with the first episode of syncope in about 30% of the patients. This finding emphasizes the importance of diagnosing LQTS in the presymptomatic period. Depending on the type of mutation present, sudden cardiac death may take place during exercise, emotional stress, at rest, or at sleep. LQT4 is associated with paroxysmal atrial fibrillation.
Studies have shown an improved response to pharmacologic treatment with a lowered rate of sudden cardiac death in LQT1 and LQT2, compared with LQT3.
Neurologic deficits after aborted cardiac arrest may complicate the clinical course of patients with LQTS after successful resuscitation.
Patient Education
Educate patients regarding the nature of long QT syndrome (LQTS) and factors that trigger cardiac events. Patients should avoid sudden noises (eg, from an alarm clock), strenuous exercise, water activities, and other arousal factors.
Educate patients and family members about the critical importance of systematic treatment with beta-blockers. Advise family members and the patient's teachers at school to undergo training in cardiopulmonary resuscitation (CPR).
Educate patients and family members about medications that may induce QT prolongation and that should be avoided in patients with LQTS. The Arizona Center for Education and Research on Therapeutics (AZCERT) provides lists of drugs that prolong the QT interval and/or induce torsade de pointes ventricular arrhythmia.
The Sudden Arrhythmia Death Syndromes Foundation (SADS) has support groups for families with LQTS.
-
Marked prolongation of QT interval in a 15-year-old male adolescent with long QT syndrome (LQTS) (R-R = 1.00 s, QT interval = 0.56 s, QT interval corrected for heart rate [QTc] = 0.56 s). Abnormal morphology of repolarization can be observed in almost every lead (ie, peaked T waves, bowing ST segment). Bradycardia is a common feature in patients with LQTS.
-
Genetically confirmed long QT syndrome (LQTS) with borderline values of QT corrected for heart rate (QTc) duration (R-R = 0.68 s, QT interval = 0.36 s, QT interval corrected for heart rate [QTc] = 0.44 s) in a 12-year-old girl. Note the abnormal morphology of the T wave (notches) in leads V2-V4.
-
ECG of a 13-year-old female who had a syncopal event while running to a school bus. She awoke after a few seconds, and her subsequent clinical course was uneventful.
Tables
Type of LQTS |
Chromosomal Locus |
Mutated Gene |
Ion Current Affected |
LQT1 |
11p15.5 |
KVLQT1 or KCNQ1 (heterozygotes) |
Potassium (IKs) |
LQT2 |
7q35-36 |
HERG, KCNH2 |
Potassium (IKr) |
LQT3 |
3p21-24 |
SCN5A |
Sodium (INa) |
LQT4 |
4q25-27 |
ANK2, ANKB |
Sodium, potassium and calcium |
LQT5 |
21q22.1-22.2 |
KCNE1 (heterozygotes) |
Potassium (IKs) |
LQT6 |
21q22.1-22.2 |
MiRP1, KNCE2 |
Potassium (IKr) |
LQT7 (Anderson syndrome) |
17q23.1-q24.2 |
KCNJ2 |
Potassium (IK1) |
LQT8 (Timothy syndrome) |
12q13.3 |
CACNA1C |
Calcium (ICa-Lalpha) |
LQT9 |
3p25.3 |
CAV3 |
Sodium (INa) |
LQT10 |
11q23.3 |
SCN4B |
Sodium (INa) |
LQT11 |
7q21-q22 |
AKAP9 |
Potassium (IKs) |
LQT12 |
|
SNTAI |
Sodium (INa) |
JLN1 |
11p15.5 |
KVLQT1 or KCNQ1 (homozygotes) |
Potassium (IKs) |
JLN2 |
21q22.1-22.2 |
KCNE1 (homozygotes) |
Potassium (IKs) |
Criterion |
Points |
|
Electrocardiogram findings * |
||
QTc, ms† |
>480 |
3 |
460-469 |
2 |
|
450-459 in male patient |
1 |
|
Torsade de pointes‡ |
2 |
|
T-wave alternans |
1 |
|
Notched T wave in 3 leads |
1 |
|
Low heart rate for age§ |
0.5 |
|
Clinical history |
||
Syncope║ |
With stress |
2 |
Without stress |
1 |
|
Congenital deafness |
0.5 |
|
Family history ¶ |
|
|
A. Family members with definite LQTS# |
1 |
|
B. Unexplained sudden cardiac death at age <30 years in an immediate family member |
0.5 |
|
LQTS = long QT syndrome. *In the absence of medications or disorders known to affect these electrocardiographic features. †QTc calculated by Bazett's formula. ‡Mutually exclusive. §Resting heart rate below the second percentile for age. ||Mutually exclusive. ¶The same family member cannot be counted in both A and B. #Definite LQTS is defined by an LQTS score above 3 (≥4). |
||
Group |
Prolonged QTc, sec |
Borderline QTc, sec |
Reference Range, sec |
Children and adolescents (<15 years) |
>0.46 |
0.44-0.46 |
<0.44 |
Men |
>0.45 |
0.43-0.45 |
<0.43 |
Women |
>0.46 |
0.45-0.46 |
<0.45 |





