Overview
Pyruvate carboxylase (PC) deficiency is a rare inborn error of metabolism that can cause developmental delay and failure to thrive starting in the neonatal or early infantile period. All patients who develop symptoms in the first weeks and months of life have lactic acidosis. PC deficiency can also start later in childhood and adulthood, with the latter presentation mimicking diabetic ketoacidosis.
PC is a biotin-dependent mitochondrial enzyme that plays an important role in energy production and anaplerotic pathways. PC catalyzes the conversion of pyruvate to oxaloacetate with biotin as a cofactor. Some cases of partial enzyme deficiency respond to the administration of excess biotin. Oxaloacetate is one of two essential substrates needed to produce citrate, the first substrate in gluconeogenesis. [1]
PC deficiency results in malfunction of the citric acid cycle and of gluconeogenesis, thereby depriving the body of energy. The citric acid cycle derives energy from carbohydrates, while gluconeogenesis produces carbohydrate fuel for the body when carbohydrate intake is low. Loss of PC activity and the citric acid (Krebs) cycle lead to decreased glutamate production, important in the nervous system. See the figure below.
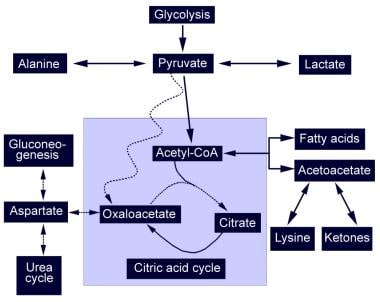 Pyruvate Carboxylase Deficiency. Diagrammatic representation of the citric acid cycle and the abnormalities found in pyruvate carboxylase deficiency (PCD). The dotted line represents absent pathways. Pyruvate cannot produce oxaloacetate and is shunted to alternative pathways that produce lactic acid and alanine. The lack of oxaloacetate limits gluconeogenesis and urea cycle function.
Pyruvate Carboxylase Deficiency. Diagrammatic representation of the citric acid cycle and the abnormalities found in pyruvate carboxylase deficiency (PCD). The dotted line represents absent pathways. Pyruvate cannot produce oxaloacetate and is shunted to alternative pathways that produce lactic acid and alanine. The lack of oxaloacetate limits gluconeogenesis and urea cycle function.
Metabolic acidosis caused by abnormal lactate production is associated with nonspecific symptoms such as moderate to severe lethargy, poor feeding, vomiting, and seizures, especially during periods of illness and metabolic stress. In the most severe form, pyruvate carboxylase deficiency results in progressive neurologic symptoms, starting in the neonatal or early infantile period, including developmental delay, poor muscle tone, abnormal eye movements, and seizures. Therapies such as alkalinization can ameliorate the biochemical abnormalities but cannot prevent the progressive neurologic damage that occurs. PC also has a role in lipogenesis in adipose tissue.
Types of pyruvate carboxylase deficiency
The following types of PC deficiency have been defined [2] :
-
Type A: The North American phenotype; characterized by infantile onset, moderate lactate elevation, normal lactate-to-pyruvate ratio, global developmental delay with intellectual disability, and survival until adulthood
-
Type B: The French phenotype; characterized by neonatal onset, high lactate and ammonia elevation, abnormal lactate-to-pyruvate ratio, and death within the first few months of life
-
Type C: The benign phenotype; characterized by recurrent episodes of mild to moderate lactate elevation without any neurologic or cognitive symptoms.
Consultations
Evaluation of the patient by an expert in metabolic and genetic disease is necessary to confirm the diagnosis, guide appropriate treatment, and determine prognosis. DNA confirmation is available at multiple laboratories as a single test or as a part of “slice” of newborn metabolic genetic disorders. Enzyme testing is not frequently done. Genetic counseling for parents is important in order to discuss the 1 in 4 recurrence risk for future pregnancies. [3]
Patient education
Patients and parents should be well educated on the factors that elicit a crisis and the early signs of decompensation. An informational statement that describes the child's disorder and the appropriate medical treatment for the disorder in an emergency setting should be carried by the patient’s parents at all times.
Pathophysiology
Pyruvate carboxylase (PC) deficiency affects metabolism in several major ways. The production of citrate, the first substrate in the citric acid cycle, is limited, thus preventing the citric acid cycle from functioning, shown in the figure below.
Pyruvate Carboxylase Deficiency. Diagrammatic representation of the citric acid cycle and the abnormalities found in pyruvate carboxylase deficiency (PCD). The dotted line represents absent pathways. Pyruvate cannot produce oxaloacetate and is shunted to alternative pathways that produce lactic acid and alanine. The lack of oxaloacetate limits gluconeogenesis and urea cycle function.
The precursor of oxaloacetate, pyruvate, is shunted toward alternate metabolic pathways, leading to an increase in lactic acid, alanine, and acetylcoenzyme A (acetyl-CoA). Acetyl-CoA cannot produce citrate without oxaloacetate and is shunted to produce ketone bodies.
Gluconeogenesis also cannot proceed without oxaloacetate, resulting in hypoglycemia during times of prolonged fasting. Tissues that are solely dependent on glucose for fuel, such as the brain, [4] are severely compromised during fasting states. Because cells cannot use the citric acid cycle to produce energy, energy is extracted from glucose exclusively through glycolysis which generates less adenosine triphosphate (ATP). High rates of glycolysis leads to hypoglycemia, thereby compounding the problem.
Aspartic acid, which is derived from oxaloacetate, is required for the urea cycle. A decrease in aspartic acid production reduces ammonia disposal and leads to increased serum ammonia levels.
PC produces intermediates of the citric acid cycle that are important for nervous system function. Alpha-ketoglutarate is a precursor for the major central nervous system excitatory neurotransmitter, glutamate. PC also has a role in producing myelin.
Etiology
The gene that encodes pyruvate carboxylase (PC) has been localized to chromosome band 11q13.2. PC deficiency is inherited from parents by an autosomal recessive inheritance pattern. Neonatal PC deficiency is associated with a complete absence of PC messenger ribonucleic acid (mRNA) and the PC enzyme protein. Infantile-onset PC deficiency is associated with residual enzyme activity that is less than 2% of normal.
In a study by Monnot et al, the authors concluded that their results, along with data already culled from previously identified PC mutations, indicated that type A PC deficiency is caused by the association of two missense mutations found in either the biotin carboxylase or carboxyltransferase (N-terminal part) domain of the PC molecule, while type B consistently arises from at least one truncating mutation, which in most cases occurs in PC's carboxyltransferase (C-terminal part) or biotin carboxyl carrier protein domain. [5] The investigators examined nine novel mutations of the PC gene in five unrelated patients, two of whom had type A and three of whom had type B PC deficiency. [5, 6]
Tsygankova et al performed an in-depth genetic analysis of four patients with PC deficiency. Two of the patients had rare genetic variants which required whole-exome sequencing/whole-genome sequencing (WES/WGS) for identification. [7] Their report added six additional variants to the mutation spectrum in PC deficiency. As of June 2022, The Human Gene Mutation Database (HGMD) listed 62 variants in the PC gene. [7]
Epidemiology
Pyruvate carboxylase (PC) deficiency is a rare disorder, with an estimated global incidence of 1 in 250,000 births. [7] Infantile-onset type A PC deficiency is more common in the United States. An increased incidence has been documented among certain populations, most notably native North Americans who speak the Algonquin dialect. [8] A founder effect has been postulated.
The neonatal onset type B pyruvate carboxylase deficiency has a greater incidence in France, although it has been described in Canada, Egypt, and Saudi Arabia.
Age of presentation for the most serious forms varies from the prenatal period to early infancy. Severe disease has prenatal onset with congenital brain abnormalities. [9] Type A cases manifest in early infancy. The benign form manifests as periods of lactic acidosis anytime during life.
Prognosis
Although dietary manipulation and supplementation of substrates and cofactors can reverse some of the biochemical abnormalities in type B PC deficiency, neurologic abnormalities progress and most patients die within the first 6 months of life. [10] The severe energy deficit in the central nervous system causes neurologic symptoms and congenital brain malformation due to a lack of energy during neurogenesis.
In neonates with apparently normal brains, progressive neurologic deterioration is variable. Hypomyelination, cystic lesions, and gliosis of the cortex or cerebellum with gray matter degeneration or necrotizing encephalopathy occur in some infants. Others develop Leigh syndrome, which is a gliosis of the brainstem and basal ganglia with capillary proliferation and characteristic changes on computed tomography (CT) and magnetic resonance imaging (MRI) scans.
Most patients with the type A pyruvate carboxylase deficiency live into adulthood but have global neurologic and cognitive dysfunction.
Patient History
Patient history in pyruvate carboxylase (PC) deficiency is characterized as follows:
-
Birth: Low Apgar scores and an infant small for dates are nonspecific signs of a metabolic disturbance during gestation
-
General: Development of poor feeding, vomiting, and lethargy are nonspecific, but common, symptoms of metabolic illnesses; symptoms can be initiated by a mild viral illness and are out of proportion to the severity of the illness
-
Developmental: Mental, psychomotor, and/or growth retardation are nonspecific signs of metabolic disease
-
Neurologic: Poor acquisition or loss of motor milestones, new-onset seizures, episodic incoordination, abnormal eye movements, and poor response to visual stimuli are signs of poor neurologic development or degenerative disease [11]
-
Respiratory: A history of apnea, dyspnea, or respiratory depression is consistent with neurologic disease or severe lactic acidosis.
-
Dermatologic: Skin may be mottled
Physical Examination
Physical examination findings in pyruvate carboxylase (PC) deficiency are neurologic, respiratory, and abdominal. Hypotonia, ataxia, tremors, and choreoathetosis are consistent with PC deficiency. Progressive motor pathway degeneration results in positive Babinski sign and spastic diplegia or quadriplegia. Ophthalmologic examination may reveal poor visual tracking, grossly disconjugate eye movements, poor pupillary response, and/or blindness. Prenatal or postnatal microcephaly also may be evident on physical examination.
Intermittent hyperpnea at rest, apnea, dyspnea, Cheyne-Stokes respiration, and respiratory failure are nonspecific signs of metabolic and neurologic disease or severe acidosis. Abdominally, hepatosplenomegaly may be appreciated.
Differential Diagnosis
Conditions to consider in the differential diagnosis of pyruvate carboxylase deficiency include the following:
-
Fatty acid beta-oxidation deficiencies
-
Leigh encephalopathy
-
Phosphoenolpyruvate carboxykinase deficiency
-
2-Ketoglutarate dehydrogenase deficiency
-
Dihydrolipoamide dehydrogenase deficiency
-
Fumarase deficiency
-
Carbonic anhydrase VA deficiency
Lab Studies and Histologic Findings
The diagnosis of PC deficiency is based on analysis of amino acids as initial diagnostic tests, followed by DNA testing in the form of direct analysis or as part of a “slice” of metabolic disorders. [3] Detection of deficiency of PC enzyme activity measured in fibroblasts is no longer widely available.
Lactate and pyruvate levels
High blood lactate and pyruvate levels with or without lactic aciduria suggests an inborn error of energy metabolism. An increased lactate-to-pyruvate ratio is characteristic of citric acid cycle disorders. This ratio may be particularly elevated during periods of crisis, such as illness or metabolic stress.
Cerebrospinal fluid shows an elevation of lactate and pyruvate. Cerebrospinal fluid glutamine is markedly reduced, while glutamic acid and proline levels are elevated.
Hypoglycemia
Hypoglycemia may occur during fasting because of reduced gluconeogenesis. The period of fasting required to produce symptoms is much shorter in pyruvate carboxylase (PC) deficiency than in other disorders.
Hyperammonemia
Hyperammonemia results from the poor ammonia disposal and decreased urea cycle function.
Absence of mRNA
The severe form of PC deficiency can be diagnosed by demonstrating the absence of PC messenger ribonucleic acid (mRNA) or specific cross-reacting material.
Histologic findings
Histologic examination of the liver may reveal lipid droplet accumulation. Central nervous system neuropathology may include poor myelination, paucity of cerebral cortex neurons, gliosis, and proliferation of astrocytes. Some patients demonstrate nemaline rods on muscle biopsy. [12]
Amino Acid Measurements
Measurement of serum amino acids may reveal the following:
-
Hyperalaninemia: Due to pyruvate shunting
-
Low aspartic acid: Due to the deficiency of the oxaloacetate precursor
-
Hypercitrullinuria and hyperlysinemia: Result from a metabolic block in the urea cycle due to low aspartic acid
Amino acid levels vary with the general metabolic state of the patient. If the patient is in a catabolic state, proteins are degraded, resulting in the elevation of many amino acids and a nonspecific amino acid profile.
MRI Findings
Type B pyruvate carboxylase (PC) deficiency is associated with ventricular dilation, cerebrocortical and white matter atrophy, or periventricular white matter cysts.
Type A is associated with symmetrical cystic lesions and gliosis in the cortex, basal ganglia, brainstem, or cerebellum and/or generalized hypomyelination, as well as hyperintensity of the subcortical frontoparietal white matter.
Magnetic resonance spectroscopy
Brain MR spectroscopy shows high lactate levels, as well as levels of N-acetylaspartate and choline consistent with hypomyelination.
Treatment
Treatments are aimed at stimulating the pyruvate dehydrogenase complex and providing alternative fuels. Correction of the biochemical abnormality can reverse some symptoms, but central nervous system damage progresses regardless of treatment. [3] Lactate levels should be closely monitored.
The pyruvate dehydrogenase complex can provide an alternative pathway for pyruvate metabolism. Pyruvate dehydrogenase complex activity can be optimized by cofactor supplementation with thiamine and lipoic acid and administration of dichloroacetate. Increased pyruvate metabolism through this pathway can help to reduce the pyruvate and lactate levels.
Other pharmacologic treatments include the following:
-
Biotin supplementation: Administered to help optimize residual enzyme activity, but it is usually of little use except potentially in a mildly affected type C patient or patient with partial enzyme function who has an increase Km of biotin cofactor binding
-
Citrate supplementation: Reduces acidosis and provides the needed substrate in the citric acid cycle
-
Aspartic acid supplementation: Allows the urea cycle to proceed and reduces the ammonia level
-
Cornstarch: One patient was reportedly treated successfully with continuous, nocturnal gastric-drip feeding with uncooked cornstarch
-
Triheptanoin: Has reportedly reversed hepatic failure and biochemical abnormalities in one case, presumably by providing a source of acetyl-CoA and anaplerotic propionyl-CoA; however, life expectancy was not prolonged and recent evidence suggests triheptanoin is ineffective [13]
-
Bezafibrate: In a case study, researchers postulated that bezafibrate may act by increasing PC mRNA expression thereby resulting in greater residual activity of the mutant PC enzyme. Although a two-fold increase of PC activity in control fibroblasts after a 72 h treatment with 400 μM bezafibrate was found, the treatment was unable to increase PC activity in a patient with PCD. [14]
Orthotopic liver transplantation has reversed the biochemical abnormalities in two patients. [3]
Diet
Diet has a small effect on outcome. A high-carbohydrate, high-protein diet may help to maintain an anabolic state and prevent activation of gluconeogenesis. [15] Fasting should be avoided. The ketogenic diet is an absolute contraindication and has been shown to worsen the acidosis to a life-threatening range. [3] A dietary log should be completed to help evaluate dietary manipulations and to ensure compliance.
Inpatient Care
Acute decompensation during illness requires admission to the hospital and management of the acidosis with hydration and intravenous bicarbonate. The patient must be supplied with adequate carbohydrates.
-
Pyruvate Carboxylase Deficiency. Diagrammatic representation of the citric acid cycle and the abnormalities found in pyruvate carboxylase deficiency (PCD). The dotted line represents absent pathways. Pyruvate cannot produce oxaloacetate and is shunted to alternative pathways that produce lactic acid and alanine. The lack of oxaloacetate limits gluconeogenesis and urea cycle function.




