Background
Diffuse parenchymal lung diseases (DPLDs) comprise a heterogenous group of disorders. Clinical, physiologic, radiographic, and pathologic presentations of patients with these disorders are varied (an example is shown in the image below). However, a number of common features justify their inclusion in a single disease category.
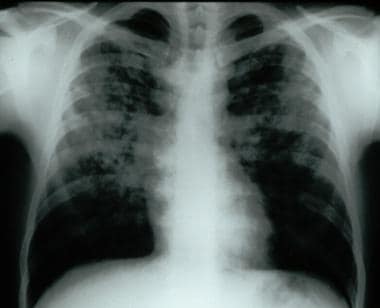 Frontal chest radiograph demonstrating bilateral reticular and nodular interstitial infiltrates with upper zone predominance.
Frontal chest radiograph demonstrating bilateral reticular and nodular interstitial infiltrates with upper zone predominance.
DPLD may be idiopathic, a classic illustration of which is idiopathic interstitial fibrosis (IPF), which is discussed in another article (see Idiopathic Pulmonary Fibrosis). The underlying histopathology of IPF is usual interstitial pneumonitis (UIP). Other major histopathologic forms of idiopathic interstitial pneumonias include the following: desquamative interstitial pneumonia (DIP), respiratory bronchiolitis interstitial lung disease (RBILD), acute interstitial pneumonitis (AIP), also known as Hamman-Rich syndrome, nonspecific interstitial pneumonia (NSIP), cryptogenic organizing pneumonia (COP) (see Imaging in Bronchiolitis Obliterans Organizing Pneumonia), and lymphocytic interstitial pneumonia (LIP) (see Lymphocytic Interstitial Pneumonia).
Some forms of DPLD are related to occupational, environmental, drug, and/or radiation exposure, as well as systemic illness such as collagen-vascular disease (see Interstitial Lung Disease Associated With Collagen-Vascular Disease). Another category of DPLDs includes granulomatous forms, such as sarcoidosis (see Sarcoidosis), and hypersensitivity pneumonia (HSP) (see Hypersensitivity Pneumonitis). Finally, a number of very rare forms of DPLDs exist, including pulmonary Langerhans cell histiocytosis (PLCH) (see Eosinophilic Granuloma (Histiocytosis X)), tuberous sclerosis, lymphangioleiomyomatosis (LAM) (see Lymphangioleiomyomatosis), and Hermansky-Pudlak syndrome.
Some of these disorders, for example, RBILD, DIP, and PLCH, are clearly associated with smoking. Some forms of DPLD, as noted above, may also be related to occupational, environmental, drug, radiation exposure, or systemic illness such as collagen-vascular disease. This article presents a broad overview, with an emphasis on those etiologies that result in pulmonary fibrosis not discussed elsewhere in this series.
Pathophysiology
A common pathophysiology has been postulated for these disorders. It is thought to begin with acute injury to the pulmonary parenchyma, leading to chronic interstitial inflammation, then to fibroblast activation and proliferation, and finally progressing to the common endpoint of pulmonary fibrosis and tissue destruction. Current research indicates that inflammation is less important in IPF, which appears to be primarily a disorder of fibroblast activation and proliferation in response to some as yet unknown trigger(s). [1]
The DPLDs typically manifest with the insidious onset of respiratory symptomatology, although onset can be acute and rapidly progressive, as in COP or AIP.
Pathologically, all DPLDs manifest histologically with disease largely within the interstitial compartment of the lung. However, alveolar and airway architecture also may be disrupted to varying degrees. The histologic patterns of UIP, DIP, nonspecific interstitial pneumonitis (NSIP), HSP, LIP, COP, giant cell pneumonitis, and granulomatous pneumonitis are most common and are focused in the alveolar, lobular, and lobar septa, impacting alveoli, small airways, and pulmonary vasculature.
Etiology
Numerous causes/diagnoses are included among the DPLDs, many of which can be grouped as shown below.
DPLDs that mimic interstitial lung disease in clinical presentation and chest radiographic findings include the following:
-
Congestive heart failure
-
Atypical pneumonia (including Pneumocystis species)
-
Lymphangitic spread of cancer
DPLDs associated with environmental or occupational exposures include the following:
-
Pneumoconioses (a group of occupational lung diseases related to inhalational exposures to inorganic dusts [eg, silicosis, asbestosis, berylliosis, coal worker's pneumoconiosis {black lung}])
-
HSP caused by exposure to protein antigens (eg, farmer's lung, pigeon-breeder's lung, hot-tub lung)
-
Fibrotic lung disease due to exposure to toxic gases, fumes, aerosols, and vapors (eg, silo-filler's disease)
-
Radiation exposure (ionizing radiation, frequently used in medical therapeutics)
DPLDs associated with rheumatologic/connective-tissue diseases include the following:
-
Scleroderma (CREST syndrome/progressive systemic scleroderma)
-
Rheumatoid arthritis
-
Mixed connective-tissue disease
-
Systemic lupus erythematosus
-
The pulmonary-renal syndromes (ie, Wegner or Goodpasture disease) - Often included with this group; however, predominant manifestation is vasculitis rather than fibrosis
DPLDs related to drug exposure include the following:
-
Cytotoxic agents (eg, bleomycin, busulfan, methotrexate)
-
Antibiotics (eg, nitrofurantoin, sulfasalazine)
-
Antiarrhythmics (eg, amiodarone, tocainide)
-
Anti-inflammatory medications (eg, gold, penicillamine)
-
Illicit drugs (eg, crack cocaine, heroin)
-
Sarcoidosis and other granulomatous diseases (eg, berylliosis)
DPLDs related to other systemic illnesses include the following:
-
Hepatitis C
-
Inflammatory bowel disease
-
Acquired immunodeficiency syndrome
Idiopathic or rare DPLDs include the following:
-
COP (idiopathic)
-
PLCH (rare)
-
Eosinophilic pneumonia
Inherited DPLDs include the following:
-
Familial IPF or sarcoidosis
-
Tuberous sclerosis
-
Neurofibromatosis
-
Niemann-Pick disease
-
Gaucher disease
-
Hermansky-Pudlak syndrome
Certain DPLDs, such as RBILD, DIP, and PLCH, are largely or only seen in current or former smokers.
Epidemiology
Frequency
United States
As a group, diffuse interstitial diseases of the lung are uncommon. Based on the Bernalillo County, NM, USA registry data published in 1994, the overall estimated incidence is approximately 30 cases per 100,000 persons per year. [2] Rates of interstitial lung disease are somewhat higher in men than in women, and the epidemiology is markedly affected by age and occupational exposures. Of patients referred to a pulmonary disease specialist, an estimated 10-15% have a DPLD.
International
Although little published data exist comparing worldwide prevalence, significant differences are apparent. The Bernalillo County study estimated a prevalence of 80.9 cases per 100,000 population in men and 67.2 cases per 100,000 population in women. In comparison, a Japanese study estimated a prevalence of 4.1 cases per 100,000 population; a study in the Czech Republic reported 7-12 cases per 100,000 population; and data from a Finnish registry indicated 16-18 cases per 100,000 population.
Race
Diffuse interstitial diseases of the lung sometimes show racial predilections. Examples include sarcoidosis, which is more common in those of African ancestry in the United States. In contrast, PLCH, also known as histiocytosis X, primarily affects Caucasians.
Sex
Several diffuse interstitial diseases of the lung show sexual predilections. IPF affects men more than women (at a ratio of 1.5:1), while LAM and pulmonary tuberous sclerosis exclusively affect women.
The Bernalillo County study estimated an incidence of 31.5 cases per 100,000/year in men and 26.1 cases per 100,000/year in women.
Women are much more likely to develop rheumatologic/connective-tissue disease than men and thus are more likely to experience pulmonary manifestations of those diseases. However, when affected, men with certain rheumatologic diseases (eg, rheumatoid arthritis) are more likely to develop pulmonary manifestations than women.
The pneumoconioses (eg, silicosis) are much more common in men than in women, probably because of higher rates of occupational exposure.
Age
Many of the DPLDs develop over many years and therefore are more prevalent in older adults. For example, most patients with IPF present in the sixth or greater decade of life. Others forms of interstitial lung disease, such as sarcoidosis, LAM, connective-tissue disease–associated lung disease, and inherited forms of lung disease primarily present in younger adults.
Prognosis
The natural history of diffuse interstitial lung diseases varies among different diagnostic entities and among individuals with the same diagnosis. Some diseases are insidious in onset and gradual but unrelenting in progression (eg, similar to IPF), while other diseases are acute in onset but responsive to therapy (eg, COP). Diseases that most closely approximate IPF have a similar mortality rate of approximately 50% at 5 years. In the United States, mortality attributed to all types of DPLDs is estimated to be 10-15% of that of chronic obstructive pulmonary disease (COPD).
Patient Education
Educate patients about the nature of the specific diagnosis and about potential toxicities of prescribed medications.
-
Frontal chest radiograph demonstrating bilateral reticular and nodular interstitial infiltrates with upper zone predominance.
-
High-resolution chest CT scan of patient with bilateral reticular and nodular interstitial infiltrates with upper zone predominance.



