Practice Essentials
Polycythemia vera (PV) is a stem cell disorder characterized as a panhyperplastic, malignant, and neoplastic marrow disorder. Its most prominent feature is an elevated absolute red blood cell mass because of uncontrolled red blood cell production. This is accompanied by increased white blood cell (myeloid) and platelet (megakaryocytic) production, which is due to an abnormal clone of the hematopoietic stem cells with increased sensitivity to the different growth factors for maturation. [1, 2, 3, 4]
Signs and symptoms of polycythemia vera
Impaired oxygen delivery due to sludging of blood may lead to the following symptoms:
-
Headache
-
Dizziness
-
Vertigo
-
Tinnitus
-
Visual disturbances
-
Angina pectoris
-
Intermittent claudication
Bleeding complications, seen in approximately 1% of patients with PV, include epistaxis, gum bleeding, ecchymoses, and gastrointestinal (GI) bleeding. Thrombotic complications (1%) include venous thrombosis or thromboembolism and an increased prevalence of stroke and other arterial thromboses.
Physical examination findings may include the following:
-
Splenomegaly (75% of patients)
-
Hepatomegaly (30%)
-
Plethora
-
Hypertension
Diagnosis of polycythemia vera
According to 2016 revised World Health Organization (WHO) guidelines, diagnosis of PV requires requires the presence of either all three major criteria or the first two major criteria and the minor criterion. [5]
Major WHO criteria are as follows:
Hemoglobin > 16.5 g/dL in men and > 16 g/dL in women, or hematocrit > 49% in men and > 48% in women, or red cell mass > 25% above mean normal predicted value
Bone marrow biopsy showing hypercellularity for age with trilineage growth (panmyelosis) including prominent erythroid, granulocytic, and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size)
Presence of JAK2V617F or JAK2 exon 12 mutation
The minor WHO criterion is as follows:
-
Serum erythropoietin level below the reference range for normal
Management of polycythemia vera
Treatment measures are as follows:
-
Phlebotomy – To keep hematocrit below 45%
-
Aspirin – 81 mg daily
-
Cytoreductive therapy – For patients at high risk for thrombosis
-
Splenectomy in patients with painful splenomegaly or repeated episodes of splenic infarction
Hydroxyurea is the most commonly used cytoreductive agent. If hydroxyurea is not effective or not tolerated, alternatives include the following:
-
Interferon alfa
-
Busulfan – In patients older than 65 years
-
Ruxolitinib (Jakafi)
-
Fedratinib (Inrebic)
For discussion of polycythemia in children, see Pediatric Polycythemia vera.
Pathophysiology
The bone marrow of patients with polycythemia vera (PV),contains normal stem cells but also contains abnormal clonal stem cells that interfere with or suppress normal stem cell growth and maturation. Evidence indicates that the etiology of panmyelosis is unregulated neoplastic proliferation. The origin of the stem cell transformation remains unknown. See the image below.
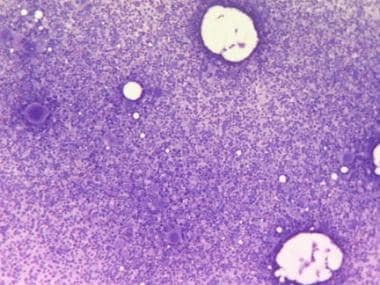 Bone marrow film at 100X magnification demonstrating hypercellularity and increased number of megakaryocytes. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Bone marrow film at 100X magnification demonstrating hypercellularity and increased number of megakaryocytes. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Progenitors of the blood cells in these patients display abnormal responses to growth factors, suggesting the presence of a defect in a signaling pathway common to different growth factors. The observation that in vitro erythroid colonies grow when no endogenous erythropoietin (Epo) is added to the culture and the presence of a truncated Epo receptor in familial erythrocytosis indicate that the defect is in the transmission of the signal. The sensitivity of PV progenitors to multiple cytokines suggests that the defect may lie in a common pathway downstream from multiple receptors. Increased expression of BCLX suggests an additional decrease in cellular apoptosis.
A mutation of the Janus kinase–2 gene (JAK2) is the most likely source of PV pathogenesis, as JAK2 is directly involved in the intracellular signaling following exposure to cytokines to which PV progenitor cells display hypersensitivity. [6] A recurrent unique acquired clonal mutation in JAK2 has been found in most patients with PV and other myeloproliferative diseases (MPDs), including essential thrombocythemia and idiopathic myelofibrosis.
A unique valine-to-phenylalanine substitution at position 617 (V617F) in the pseudokinase JAK2 domain has been identified. The substitution, called JAK2V617F, leads to a permanently turned-on signaling at the affected cytokine receptors. [7, 8, 9, 10] The JAK2V617F mutation is present in more than 95% of PV cases, but is also found in 50%-60% of essential thrombocytosis and primary myelofibrosis cases. [11] How these mutations interact with the wild-type kinase genes and how they manifest into different forms of MPDs need to be elucidated.
At diagnosis of PV, a homozygous JAK2 genotype is found less often in women than in men (median, 61% vs 80%). JAK2 variant allele frequency, which is initially similar in men and women, over time becomes significantly higher in men than in women. [12]
Thrombosis and bleeding are frequent in persons with PV, as a result of the disruption of hemostatic mechanisms because of (1) an increased level of red blood cells and (2) an elevation of the platelet count. There are findings that indicate the additional roles of tissue factor and polymorphonuclear leukocytes (PMLs) in clotting, the platelet surface as a contributor to phospholipid-dependent coagulation reactions, and the entity of platelet microparticles. Tissue factor is also synthesized by blood leukocytes, the level of which is increased in persons with MPD, which can contribute to thrombosis.
PV tends to be milder in women than in men, with lower rates of myocardial infarction and peripheral arterial disease (although this may be related to lower rates of smoking in women). However, venous thrombosis is more common in females; in particular, the rate of splanchnic vein thrombosis is significantly higher in young women. [12]
Rusak et al evaluated the hemostatic balance in patients using thromboelastography and also studied the effect of isovolemic erythrocytapheresis on patients with polycythemia vera. They concluded that thromboelastography may help to assess the thrombotic risk in patients with polycythemia vera. [13]
Hyperhomocystinemia is a risk factor for thrombosis and is also widely prevalent in patients with MPD (35% in controls, 56% in persons with polycythemia vera).
Acquired von Willebrand syndrome is an established cause of bleeding in persons with MPD, accounting for approximately 12-15% of all patients with this syndrome. von Willebrand syndrome is largely related to the absorption of von Willebrand factor onto the platelets; reducing the platelet count should alleviate the bleeding from the syndrome.
Epidemiology
Frequency
United States
Polycythemia vera (PV) is relatively rare, occurring in 0.6-1.6 persons per million population.
Race-, Sex-, and Age-related Demographics
Originally, Ashkenazi Jewish persons were thought to have a higher predilection for polycythemia vera than members of other ethnic groups. Subsequently, however, many studies have shown that this condition occurs in all ethnic groups.
Most studies have found the incidence of polycythemia vera to be slightly higher in males than females. However, a slightly higher incidence in females has also been reported, and one systematic meta-analysis showed no significant difference in the the crude annual incidence between males and females. [12]
The peak incidence of polycythemia vera is age 50-70 years. However, this condition occurs in persons of all age groups, including early adulthood and childhood, albeit rarely.
-
Bone marrow film at 100X magnification demonstrating hypercellularity and increased number of megakaryocytes. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Blood film at 400X magnification demonstrating polyglobulia and thrombocytosis. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
Bone marrow film at 400X magnification demonstrating dominance of erythropoiesis. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
-
This blood film at 10,000X magnification shows a giant platelet and an eosinophil. Erythrocytes show signs of hypochromia as a result of repeated phlebotomies. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.