Practice Essentials
Classic polyarteritis nodosa (PAN or c-PAN) is a systemic vasculitis characterized by necrotizing inflammatory lesions that affect medium-sized and small muscular arteries, preferentially at vessel bifurcations. [1] These lesions result in microaneurysm formation, aneurysmal rupture with hemorrhage, thrombosis, and, consequently, organ ischemia or infarction. [2]
Kussmaul and Maier first described PAN in 1866. The autopsy of a patient with fever, weight loss, abdominal pain, and polyneuropathy revealed areas of focal inflammatory exudations that gave rise to palpable nodules along the course of medium-sized arteries. [3]
PAN, like other vasculitides, affects multiple systems and has protean manifestations, although it most commonly affects skin (see the image below), joints, peripheral nerves, the gut, and the kidney. [4] The lungs are usually spared with PAN. A typical PAN patient might present with fever, night sweats, weight loss, skin ulcerations or tender nodules, and severe muscle and joint pains developing over weeks or months. (See Etiology, Presentation, and Workup.)
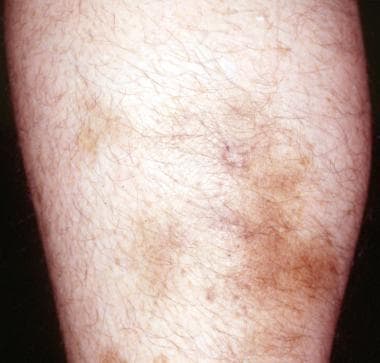 Nonspecific, firm, tender subcutaneous nodules without livedo reticularis and/or systemic involvement may be the first sign of polyarteritis nodosa (PAN).
Nonspecific, firm, tender subcutaneous nodules without livedo reticularis and/or systemic involvement may be the first sign of polyarteritis nodosa (PAN).
See Cutaneous Clues to Accurately Diagnosing Rheumatologic Disease, a Critical Images slideshow, to help recognize cutaneous manifestations of rheumatologic diseases. Also see the slideshow Vasculitis: Case Presentations for more information on clinical, histologic, and radiographic imaging findings in various forms of vasculitis. For information on pediatric PAN, see Childhood Polyarteritis Nodosa.
Insight into PAN requires some understanding of how this rare disease has been defined. Periarteritis nodosa was a term used from the mid-1800s to the 1900s to describe a spectrum of systemic vasculitic disorders, including diseases that manifested as arterial aneurysms, as well as those that caused diffuse necrotizing glomerulonephritis. [5, 6] The term periarteritis nodosa was changed to polyarteritis nodosa in the mid-1900s to reflect the transmural inflammation of arteries caused by this disorder. [7]
The understanding of vasculitides continued to increase by the 1980s with the discovery of antineutrophil cytoplasmic antibodies (ANCAs). Microscopic polyangiitis (MPA; formerly called microscopic polyarteritis) is an ANCA-associated systemic vasculitis that has some features similar to those of classic PAN, with the additional involvement of renal glomeruli and pulmonary capillaries.
Features of PAN
In 1990, the American College of Rheumatology (ACR) established criteria for research purposes in order to differentiate PAN from other forms of vasculitis. [8] A committee of ACR physicians selected 10 disease features of PAN; in order for PAN to be diagnosed, at least 3 of the 10 ACR criteria should be present when a radiographic or pathological diagnosis of vasculitis is made [8] (see Presentation and Workup):
-
Weight loss of 4 kg or more
-
Livedo reticularis
-
Testicular pain/tenderness
-
Myalgia or leg weakness/tenderness
-
Mononeuropathy or polyneuropathy
-
Diastolic blood pressure greater than 90 mm/Hg
-
Elevated blood urea nitrogen (BUN) or creatinine level unrelated to dehydration or obstruction
-
Presence of hepatitis B surface antigen or antibody in serum
-
Arteriogram demonstrating aneurysms or occlusions of the visceral arteries
-
Presence of polymorphonuclear neutrophils in a biopsy specimen from a small- or medium-sized artery
The strong association of MPA with ANCA, as well as the pathologic and clinical differences between MPA and PAN, demonstrate that PAN and MPA are likely separate disorders. It was not until 1994 that histologic criteria to distinguish PAN from MPA were defined at the international Chapel Hill Consensus Conference (CHCC). [9] According to the CHCC criteria, the presence of vasculitis in arterioles, venules, and capillaries defines the diagnosis of MPA (although small- and medium-sized arteries may also be involved in MPA) and excludes the diagnosis of PAN. (See Presentation, DDx, and Workup.)
Stages
PAN is divided into subacute, acute, and chronic stages. In the subacute stage, infiltration of mononuclear cells becomes more prominent, while in the acute stage, polymorphonuclear neutrophils infiltrate all layers of the vessel wall. (See Etiology.)
In the chronic stage, fibrinoid necrosis of the vessels causes thrombosis and tissue infarction. Aneurysmal dilatations of the involved arteries, as large as 1 cm in size, are characteristic findings of PAN. Kidney lesions show predominant arteritis without glomerulonephritis; however, in patients with severe hypertension, glomerulosclerosis may be superimposed with glomerulonephritis. Pulmonary arteries are not involved, and bronchial artery involvement is uncommon.
Treatment
Corticosteroids are the cornerstone of treatment. Additional courses include the following:
-
Idiopathic PAN that is steroid refractory or includes major organ involvement: corticosteroids plus cyclophosphamide are the standard of care
-
Hepatitis B–related PAN: corticosteroids with antiviral agents (eg, vidarabine, interferon alpha-2b) and plasmapheresis
-
In patients with steroid-refractory and recurrent PAN, case reports describe response to treatment with biologic agents (eg, infliximab, etanercept, tocilizumab, tofacitinib, rituximab)
-
Severe PAN: plasma exchange has been used
See Treatment and Medication.
For patient education information, see What Is Polyarteritis Nodosa?.
Pathophysiology
Polyarteritis nodosa (PAN) spares large vessels (the aorta and its major branches), the smallest vessels (capillaries and small arterioles), and the venous system. [7] Vascular lesions affect medium-sized muscular arteries and occur mainly at bifurcations and branch points.
Inflammation may start in the vessel intima and progress to include the entire arterial wall, destroying the internal and external elastic lamina, resulting in fibrinoid necrosis. [7] Aneurysms develop in the weakened vessel, carrying a subsequent risk for rupture and hemorrhage. Thrombi may develop at the site of the lesions. As lesions progress, proliferation of the intima or media may result in obstruction and subsequent tissue ischemia or infarction. [10]
Etiology
Hepatitis B and PAN
The pathogenesis of polyarteritis nodosa (PAN) is unknown, and no animal model is available for study. Hepatitis B virus (HBV) infection is strongly linked with PAN. Evidence for immune complex–induced disease is confined to HBV-related PAN; the role of immune complexes in non-HBV-related PAN remains unclear. [7]
Impaired function of endothelial cells may be part of idiopathic PAN or a consequence of it; in HBV-PAN, virus replication may directly injure the vessel wall. [11] Endothelial dysfunction can perpetuate the inflammation through cytokine and adhesion molecule production. [10]
HBV-associated vasculitis almost always takes the form of PAN. HBV-PAN may occur at any time during the course of acute or chronic hepatitis B infection, although it typically occurs within 6 months of infection. [11]
The activity of HBV-PAN does not parallel that of the hepatitis, and symptoms are the same as those of idiopathic PAN. Small studies have found that gastrointestinal manifestations, malignant hypertension, renal infarction, and orchiepididymitis were more common in HBV-PAN. [11]
HBV was once the cause of up to 30% of PAN cases. [12] Widespread use of the hepatitis B vaccine has significantly decreased the incidence of HBV-PAN, which is now estimated to account for less than 8% of all PAN cases. [13]
Genetic associations
Loss-of-function mutations in CECR1 (alsoknown as ADA2), the gene that encodes adenosine deaminase 2 (ADA2), have been associated with a spectrum of vascular and inflammatory phenotypes that includes polyarteritis nodosa. [14] Navon Elkan and colleagues identified six families with multiple cases of systemic and cutaneous polyarteritis nodosa, most of which had onset during childhood. In all the families, disease was traced to recessive mutations in CECR1 that resulted in reduced activity of ADA2. [15]
Possible roles of ADA2 include regulation of the proliferation of activated T cells and macrophages and the differentiation of monocytes to macrophages. Reduction in ADA2 activity may affect the adenosine inflammatory-response pathway. [15]
Similarly, Gonzalez Santiago et al report two siblings with novel compound heterozygous mutations in CECR1 who were diagnosed with cutaneous PAN in early childhood. [16]
In a study of patients with early-onset livedo reticularis and/or hemorrhagic/ischemic strokes in the context of inflammation or PAN, Caorsi et al detected biallelic homozygous or compound heterozygous CECR1 mutations in 15 of 48 patients from 43 families. In patients with CECR1 mutations, the mean age of onset of disease was 24 months (6 months to 7 years). [17]
Other disease associations
Controversy has surrounded the potential association of hepatitis C virus (HCV) with PAN. HCV may be linked to cutaneous PAN, a benign, limited form of PAN. In a study of 16 patients with cutaneous PAN, 5 tested positive for hepatitis C. [18] HCV-associated PAN has also been described in 31 patients included in a larger 161 patient cohort with HCV-related vasculitis in France. [19] Despite the presence of serum cryoglobulins, these patients were diagnosed with HCV-PAN on the basis of typical histopathologic features of PAN and/or the presence of microaneurysms and/or multiple stenoses on abdominal and/or renal angiography.
A number of other infectious organisms have been reported in association with PAN or PAN-like diseases, but causal evidence is inconsistent. These organisms include varicella-zoster virus, parvovirus B-19, cytomegalovirus, human T-cell leukemia virus, streptococcal species, Klebsiella species, Pseudomonas species, Yersinia species, Toxoplasma gondii, Rickettsiae, trichinosis, and sarcosporidiosis. [20, 21] Recently, reports of associations with PAN and human immunodeficiency virus [22] and cutaneous PAN and tuberculosis [23] have been published as well.
Some syndromes, including rheumatic diseases, malignancies, and infections have been associated with clinical syndromes indistinguishable from idiopathic PAN. Rheumatoid arthritis (RA) and Sjögren syndrome have been associated with PAN. Notably, the incidence of RA-associated vasculitis has decreased greatly since the 1980s, likely attributable to improvements in the management of RA. [24] Cutaneous PAN occurring with HLA-B39 spondyloarthritis, [25] common variable immunodeficiency, [26] and psoriatic arthritis in an 11-year-old boy [27] have also been reported.
Hematologic malignancies, such as hairy cell leukemia and, in one case, angioimmunoblastic T cell lymphoma, have been associated with PAN-like vasculitides. [28, 29]
Epidemiology
Occurrence in the United States
Polyarteritis nodosa (PAN) is a rare disease, with an incidence of about 3-4.5 cases per 100,000 population annually. Older estimates placed the prevalence as high as 7.7 cases per 100,000 population, for example, in a population of Alaskan Eskimos hyperendemic for HBV infection. [30]
International occurrence
Depending on the definitions used, the annual estimated incidence of PAN ranges from 1.6 cases per million in south Sweden to 4.6 cases per million in England to 30.7 cases per million adults in Paris, France. [12, 31]
Sex- and age-related demographics
PAN affects men more frequently than women (male-to-female ratio 1.6-2:1). PAN has been diagnosed in persons of every age; however, it is predominantly observed in individuals aged approximately 45-65 years. [10]
Prognosis
Idiopathic (non–HBV-related) PAN
Traditionally, it has been taught that relapses of polyarteritis nodosa (PAN) are rare in individuals who completely recover. However, a study in Sweden described 10 patients with PAN, 57% of whom experienced relapse within 5 years. [31]
Recovery from neurologic deficits due to PAN can take up to 18 months. Central nervous system (CNS) involvement carries a worse prognosis than does peripheral nerve involvement. [20]
The prognosis is markedly worse in patients with acute abdominal syndromes characterized by extensive bowel involvement. [32] Multiple perforations may be found, relapses are common, and the postoperative course is complicated by infections and delayed healing. Surgery performed for cholecystitis or appendicitis does not appear to worsen prognosis in the same way.
The prognosis is better in patients with cutaneous PAN without systemic involvement. This disease is benign but tends to relapse. Kato et al reported higher risk of relapse in patients with cutaneous PAN who had pretreatment cutaneous ulcers, elevated C-reactive protein level, higher absolute neutrophil count (> 4.9 × 103/μL) and neutrophil-to-lymphocyte ratio, and higher systemic immune-inflammation index. [33]
In a retrospective study of 52 patients with childhood-onset PAN who were followed for a mean of 6.2 years, 27 patients (51.9%) were in clinical remission without medication at follow-up, 17 (32.7%) were in clinical remission while on medication, and six patients (11.6%) had a persistent or relapsing disease course. Two patients (3.8%) with severe cerebral involvement died. Cranial nerve palsy occurring during the course of disease was significantly correlated with a worse prognosis. Nephrogenic hypertension at disease onset and seizures during the course of the disease were significantly associated with irreversible organ damage. [34]
Relapses were more frequent among PAN patients with severe gastrointestinal involvement in a retrospective study of 69 pediatric patients, while a higher cumulative dose of cyclophosphamide was associated with a lower relapse risk. During follow-up with a median duration of 6 years, the relapse rate was 35%. The mortality rate was 4%. [35]
HBV-related PAN
Patients who seroconvert usually recover. Once HBV-PAN goes into remission, the risk of recurrence is very low (6% in one series). [13]
HCV-related PAN
One study found that in patients with HCV-related vasculitis, HCV-PAN exhibits a more severe clinical presentation but a higher rate of clinical remission. [19]
Complications
Permanent morbidity due to PAN is relatively rare, although patients may develop peripheral neuropathy, renal insufficiency or renal failure, and/or hypertension. Fever, weight loss, and malaise are present in 50% of patients; renal failure and hypertension, in 60%; arthritis, arthralgia, and myalgia, in 64%; and peripheral neuropathy and mononeuritis multiplex, in 51%.
Complications of PAN include the following:
-
Cutaneous ulcerations
-
Extremity gangrene [36]
-
Organ infarction
-
Stroke
-
Encephalopathy
-
Myelopathy
-
Heart failure
-
Myocardial infarction/li>
-
Pericarditis
-
Renal failure
-
Gastrointestinal (GI) bleeding
-
Bowel infarction
-
Peripheral neuropathy
Mortality
When left untreated, the 5-year survival rate of PAN is 13%. Nearly half of patients die within the first 3 months of onset. Corticosteroid treatment improves the 5-year survival rate to 50-60%. When the steroid is combined with other immunosuppressants, the 5-year survival rate may increase to greater than 80%.
Death associated with PAN occurs as a result of uncontrolled vasculitis, infectious complications related to treatment-induced immunosuppression, and vascular complications of the disease, such as myocardial infarction and stroke. The mortality rate is higher in patients with acute abdominal syndromes. [32] Intractable hypertension contributes to morbidity and mortality rates.
In a cohort of 161 patients with HCV-related vasculitis, there was no significant difference in mortality between HCV-MC patients and those with HCV-PAN. Overall survival rates at 1, 3, and 5 years in HCV-PAN patients were 95% (95% confidence interval [CI], 0.87-1), 89% (95% CI, 0.75-0.98), and 89% (95% CI, 0.75-1), respectively. [19]
Prognostic score
In a prospective study of 342 patients with PAN, Guillevin et al found 5 factors associated with poor prognosis. They devised a 5-factors score (FFS) to predict survival and help guide treatment decisions. [43] The presence of any of the following 5 factors predicts an increased likelihood of mortality:
-
Renal insufficiency (serum creatinine >1.58 mg/dL)>
-
Proteinuria (>1 g/d)
-
GI involvement (bleeding, perforation, infarction, pancreatitis)
-
Cardiomyopathy
-
CNS involvement
When the FFS is zero, the predicted mortality rate at 5 years is 11.9%. When the FFS is 1, the mortality rate is 25.9%, and when the FFS is 2 or more, the mortality rate is 45.9%.
Patient Education
Patients should understand that PAN can be a progressive systemic disease, and further complications and the involvement of other organ systems are quite common. Many patients attempt to discontinue their medications after initial symptomatic improvement, owing to the potential for adverse effects. Therefore, the benefits of medical treatments should be discussed clearly with the patient, in addition to the risks associated with the long-term use of immunosuppressants. The use of these medications necessitates close monitoring for many years to come.
For patient education information, see What Is Polyarteritis Nodosa?.
-
Nonspecific, firm, tender subcutaneous nodules without livedo reticularis and/or systemic involvement may be the first sign of polyarteritis nodosa (PAN).
-
Tender, hyperpigmented, firm subcutaneous nodules with a background of livedo reticularis common in cutaneous polyarteritis nodosa (PAN).
-
Tender erythematous nodules with central "punched out" ulcerations common in cutaneous polyarteritis nodosa (PAN).
-
Polyarteritis nodosa (PAN) is characterized by fibrinoid necrosis of the arterial wall with a leukocytic infiltrate. In this slide, a large, pale occlusion of a muscular artery can be seen. Within this collagenous tissue is a leukocytic infiltrate, which is the hallmark of PAN. Courtesy of Urbana Atlas of Pathology.
-
Livedo reticularis in polyarteritis nodosa (PAN).







