Practice Essentials
A pheochromocytoma (see the image below) is a rare, catecholamine-secreting tumor derived from chromaffin cells. The term pheochromocytoma (in Greek, phios means dusky, chroma means color, and cytoma means tumor) refers to the color the tumor cells acquire when stained with chromium salts.
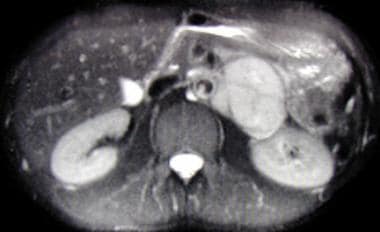 Axial, T2-weighted magnetic resonance imaging (MRI) scan showing large left suprarenal mass of high signal intensity on a T2-weighted image. The mass is a pheochromocytoma.
Axial, T2-weighted magnetic resonance imaging (MRI) scan showing large left suprarenal mass of high signal intensity on a T2-weighted image. The mass is a pheochromocytoma.
About 30% of pheochromocytomas occur as part of hereditary syndromes. Although pheochromocytomas have classically been associated with 3 syndromes—von Hippel-Lindau (VHL) syndrome, multiple endocrine neoplasia type 2 (MEN 2), and neurofibromatosis type 1 (NF1)—there are now 10 genes that have been identified as sites of mutations leading to these tumors. These different genes produce pheochromocytomas with different ages of onset, secretory profiles, locations, and potential for malignancy. [1]
Because of excessive catecholamine secretion, pheochromocytomas may precipitate life-threatening hypertension or cardiac arrhythmias. [6] If the diagnosis of a pheochromocytoma is overlooked, the consequences can be disastrous, even fatal; however, if a pheochromocytoma is found, it is potentially curable. (See Pathophysiology, Prognosis, and Treatment.) [2]
About 85% of pheochromocytomas are located within the adrenal glands, and 98% are within the abdomen. When such tumors arise outside of the adrenal gland, they are termed extra-adrenal pheochromocytomas, or paragangliomas.
Extra-adrenal pheochromocytomas develop in the paraganglion chromaffin tissue of the nervous system. They may occur anywhere from the base of the brain to the urinary bladder. Common locations for extra-adrenal pheochromocytomas include the organ of Zuckerkandl (close to the origin of the inferior mesenteric artery), bladder wall, heart, mediastinum, and carotid and glomus jugulare bodies. (See Workup.)
Malignancy
Approximately 10% of pheochromocytomas and 35% of extra-adrenal pheochromocytomas are malignant. Only the presence of metastases defines malignancy. However, specific histologic features help to differentiate adrenal pheochromocytomas with a potential for biologically aggressive behavior from those that behave in a benign fashion. Among the features that suggest a malignant course are large tumor size and an abnormal DNA ploidy pattern (aneuploidy, tetraploidy). [3] Common metastatic sites include bone, liver, and lymph nodes.
For discussion of pheochromocytoma in children, see the Medscape Drugs & Diseases article Pediatric Pheochromocytoma. [4]
Signs and symptoms of pheochromocytoma
Classically, pheochromocytoma manifests as spells with the following 4 characteristics:
-
Headaches
-
Palpitations
-
Diaphoresis
-
Severe hypertension
Typical patterns of the spells are as follows:
-
Frequency may vary from monthly to several times per day
-
Duration may vary from seconds to hours
-
Over time, spells tend to occur more frequently and become more severe as the tumor grows
The following may also occur during spells:
-
Tremor
-
Nausea
-
Weakness
-
Anxiety, sense of doom
-
Epigastric pain
-
Flank pain
-
Constipation
Clinical signs associated with pheochromocytomas include the following:
-
Hypertension: Paroxysmal in 50% of cases
-
Postural hypotension: From volume contraction
-
Hypertensive retinopathy
-
Weight loss
-
Pallor
-
Fever
-
Tremor
-
Neurofibromas
-
Tachyarrhythmias
-
Pulmonary edema
-
Cardiomyopathy
-
Ileus
-
Café au lait spots
See Clinical Presentation for more detail.
Diagnosis of pheochromocytoma
Diagnostic tests for pheochromocytoma include the following:
-
Plasma metanephrine testing: 96% sensitivity, 85% specificity [2]
-
24-hour urinary collection for catecholamines and metanephrines: 87.5% sensitivity, 99.7% specificity [5]
Test selection criteria include the following:
-
Use plasma metanephrine testing in patients at high risk (ie, those with predisposing genetic syndromes or a family or personal history of pheochromocytoma)
-
Use 24-hour urinary collection for catecholamines and metanephrines in patients at lower risk
Imaging studies should be performed only after biochemical studies have confirmed the diagnosis of pheochromocytoma. Studies are as follows:
-
Abdominal CT scanning: Has accuracy of 85-95% for detecting adrenal masses with a spatial resolution of 1 cm or greater
-
MRI: Preferred over CT scanning in children and pregnant or lactating women; has reported sensitivity of up to 100% in detecting adrenal pheochromocytomas
-
Scintigraphy: Reserved for biochemically confirmed cases in which CT scanning or MRI does not show a tumor
-
PET scanning: A promising technique for detection and localization of pheochromocytomas
Additional studies to rule out a familial syndrome in patients with confirmed pheochromocytoma include the following:
-
Serum intact parathyroid hormone level and a simultaneous serum calcium level to rule out primary hyperparathyroidism (which occurs in MEN 2A)
-
Screening for mutations in the ret proto-oncogene (which give rise to MEN 2A and 2B) [6]
-
Genetic testing for mutations causing the MEN 2A and 2B syndromes
-
Consultation with an ophthalmologist to rule out retinal angiomas (VHL disease)
See Workup for more detail.
Management of pheochromocytoma
Surgical resection of the tumor is the treatment of choice and usually cures the hypertension. Careful preoperative treatment with alpha and beta blockers is required to control blood pressure and prevent intraoperative hypertensive crises. [7]
Preoperative medical stabilization is provided as follows:
-
Start alpha blockade with phenoxybenzamine 7-10 days preoperatively
-
Provide volume expansion with isotonic sodium chloride solution
-
Encourage liberal salt intake
-
Initiate a beta blocker only after adequate alpha blockade, to avoid precipitating a hypertensive crisis from unopposed alpha stimulation
-
Administer the last doses of oral alpha and beta blockers on the morning of surgery
See Treatment and Medication for more detail.
Pathophysiology
The clinical manifestations of a pheochromocytoma result from excessive catecholamine secretion by the tumor. Secretion may occur either intermittently or continuously. Catecholamines typically secreted are norepinephrine and epinephrine; some tumors produce dopamine. [8]
The biologic effects of catecholamines are well known. Stimulation of alpha-adrenergic receptors results in elevated blood pressure, increased cardiac contractility, glycogenolysis, gluconeogenesis, and intestinal relaxation. Stimulation of beta-adrenergic receptors results in an increase in heart rate and contractility. [7]
Catecholamine secretion in pheochromocytomas is not regulated in the same manner as in healthy adrenal tissue. Unlike the healthy adrenal medulla, pheochromocytomas are not innervated, and catecholamine release is not precipitated by neural stimulation. The trigger for catecholamine release is unclear, but multiple mechanisms have been postulated, including direct pressure, medications, and changes in tumor blood flow.
Relative catecholamine levels also differ in pheochromocytomas. Most pheochromocytomas secrete norepinephrine predominantly, whereas secretions from the normal adrenal medulla are roughly 85% epinephrine.
In a study using cardiac magnetic resonance imaging, Ferreira et al found that patients with pheochromocytoma who underwent curative surgery nonetheless continued to demonstrate systolic and diastolic strain, focal fibrosis, and T1 abnormalities, with the last possibly indicating the presence of diffuse fibrosis. According to the investigators, the study’s results suggest more than just hypertensive heart disease at work and that catecholamine toxicity in pheochromocytoma may be responsible for long-lasting myocardial changes. [9]
In hereditary forms of pheochromocytoma, the secretory profiles vary according to the underlying syndrome. Eisenhofer et al found that pheochromocytomas associated with VHL typically produce norepinephrine only, while those associated with MEN 2 and NF1 typically produce both epinephrine and norepinephrine. Tumors in patients with germline mutations of succinate dehydrogenase subunit genes (SDHB and SDHD), which cause familial paraganglioma, principally produce dopamine. [10]
Precipitants of hypertensive crisis
Precipitants of a hypertensive crisis include the following:
Etiology
Although the majority of pheochromocytomas are sporadic, approximately 30% result from inherited mutations. To date, 10 genes associated with pheochromocytoma and paraganglioma have been identified. [1] Familial syndromes associated with pheochromocytomas include MEN 2A and 2B, neurofibromatosis (von Recklinghausen disease), and VHL disease, as well as others.
MEN 2
The MEN 2A and 2B syndromes have been traced to germline mutations in the ret proto-oncogene on chromosome 10, which encodes a tyrosine kinase receptor involved in the regulation of cell growth and differentiation. Pheochromocytomas occur bilaterally in the MEN syndromes in as many as 70% of cases.
MEN 2A
MEN 2A (Sipple syndrome) is characterized by the following:
-
Medullary thyroid carcinoma
-
Parathyroid adenoma
-
Pheochromocytomas
-
Hirschsprung disease.
Over 95% of cases of MEN 2A are associated with mutations in the ret proto-oncogene affecting 1 of 5 codons, located in exon 10 (codons 609, 611, 618, 620) and exon 11 (codon 634).
Clinical diagnosis of MEN 2A requires the occurrence of 2 or more endocrine tumors in one individual or in close relatives. The risk for medullary thyroid carcinoma is 95%, the risk for pheochromocytoma is 50%, and the risk for parathyroid disease is between 20% and 30%. [12]
MEN 2B
MEN 2B is characterized by the following:
-
Medullary thyroid carcinoma
-
Pheochromocytoma
-
Mucosal neurofibromatosis
-
Intestinal ganglioneuromatosis
-
Hirschsprung disease
-
Marfanoid body habitus
Patients with MEN 2B may also have ganglioneuromatosis of the gastrointestinal (GI) tract, which can cause functional GI problems. A germline missense mutation in the tyrosine kinase domain of the ret proto-oncogene (exon 16, codon 918) has been reported to be present in 95% of patients with MEN 2B.
Clinical diagnosis of MEN 2B is based on the presence of mucosal neuromas of the oral mucosa, enlarged lips with characteristic facial appearance, and marfanoid habitus. Medullary thyroid carcinoma is virtually assured with MEN 2B, and the risk of pheochromocytoma is 50%. Parathyroid disease is uncommon with MEN 2B. [12] Persons diagnosed with MEN 2B should have a prophylactic thyroidectomy in infancy because of the early and aggressive nature of associated medullary thyroid carcinoma.
Other mutational etiologies
Novel mutations that cause hereditary pheochromocytoma have been identified in the MYC-associated factor X (MAX) gene. Loss of MAX function is correlated with metastatic potential. [13] Burnichon et al concluded that germline mutations in MAX are responsible for approximately 1% of pheochromocytomas and paragangliomas in patients without evidence of other known mutations. [14]
A number of other genes, such as the GDNF gene, are associated with development of adrenal or extra-adrenal pheochromocytomas. The GDNF gene is also associated with central hypoventilation syndrome and susceptibility to Hirschsprung disease.
The TMEM127 gene also is associated with susceptibility to pheochromocytoma. Several families have been described with unique mutations to this gene that have resulted in the development of pheochromocytoma between young adulthood and middle age. All of these are inherited in an autosomal dominant fashion with incomplete penetrance.
VHL disease
VHL disease is associated with the following:
-
Pheochromocytoma
-
Cerebellar hemangioblastoma
-
Renal cell carcinoma
-
Renal and pancreatic cysts
-
Epididymal cystadenomas
One study found that this syndrome was present in nearly 19% of patients with pheochromocytomas. [15]
VHL disease is caused by mutations in the VHL gene. [16] This gene encodes a protein that plays a role in cilia formation, regulation of cellular senescence, and the oxygen-sensing pathway.
Neurofibromatosis and other diseases
Neurofibromatosis, or von Recklinghausen disease, is characterized by congenital anomalies (often benign tumors) of the skin, nervous system, bones, and endocrine glands. Only 1% of patients with neurofibromatosis have been found to have pheochromocytomas, but as many as 5% of patients with pheochromocytomas have been found to have neurofibromatosis.
Other neuroectodermal disorders associated with pheochromocytomas include tuberous sclerosis (Bourneville disease, epiloia) and Sturge-Weber syndrome.
Pheochromocytomas may produce calcitonin, opioid peptides, somatostatin, corticotropin, and vasoactive intestinal peptide. Corticotropin hypersecretion has caused Cushing syndrome, and vasoactive intestinal peptide overproduction causes watery diarrhea.
Succinate dehydrogenase complex
The succinate dehydrogenase complex subunit D protein is encoded by the SDHD gene, mutations in which cause pheochromocytomas, paragangliomas, and other tumors. In most tumors, inheritance of the mutation is autosomal dominant with biallelic expression of the SDHD gene. However, paternal imprinting appears to be the inheritance pattern in paragangliomas and, in particular, carotid body tumors resulting from the SDHD gene.
The succinate dehydrogenase complex subunit B protein is encoded by the SDHB gene. Mutations in this gene are known to cause carotid body tumors and paragangliomas and are inherited in an autosomal dominant fashion. Paragangliomas caused by SDHB mutations have a higher rate of malignant transformation that those that are not.
The succinate dehydrogenase subunit C protein is encoded by the SDHC gene, and mutations are known to cause paraganglioma. One family with a mutation in this gene showed maternal inheritance of the condition, [17] but subsequent investigation has suggested an autosomal dominant inheritance pattern without evidence of imprinting.
Other succinate dehydrogenase subunit genes with mutations linked to paraganglioma include SDHA [18] and the newly characterized succinate dehydrogenase complex assembly factor 2 (SDHAF2) gene. [19] Kunst et al found phenotypic expression of the SDHAF2 mutation only with paternal inheritance, which suggests imprinting of the gene. [19]
Hemihyperplasia
Although its genetics remain incompletely understood, hemihyperplasia (also called hemihypertrophy) is known to increase tumor risk. The condition may be an isolated finding or a part of a larger syndrome such as Beckwith-Wiedemann syndrome, Proteus syndrome, or neurofibromatosis. The tumors most commonly associated with hemihyperplasia are Wilms tumor and hepatoblastoma, but at least one patient has been described with isolated hemihyperplasia and an adrenal pheochromocytoma on the hyperplastic side. [20]
Hemihyperplasia can be caused by paternal uniparental disomy for the 11p15 chromosomal region, as can be seen in isolated hemihyperplasia and Beckwith-Wiedemann syndrome. Methylation of the LIT1 and H19 genes is important to the pathogenesis of hemihyperplasia and underscores the importance of epigenetics in normal growth and in the development of neoplasia.
Epidemiology
Pheochromocytomas are rare, reportedly occurring in 0.05–0.2% of hypertensive individuals. This accounts for only a portion of cases, however, as patients may be completely asymptomatic. A retrospective study from the Mayo Clinic revealed that in 50% of cases of pheochromocytoma, the diagnosis was made at autopsy. [21] Approximately 10% of pheochromocytomas are discovered incidentally. [22]
A Dutch study, by Berends et al, found an increase in the age-standardized incidence rate (ASR) of pheochromocytomas and sympathetic paragangliomas in the Netherlands between 1995 and 2015. The investigators reported that the ASR between 1995 and 1999 was 0.29 per 100,000 person-years, compared with 0.46 per 100,000 person-years between 2011 and 2015. The ASRs for sympathetic paragangliomas rose between these same two periods from 0.08 to 0.11 per 100,000 person-years. There was also a trend during this 20-year period towards patients being older and tumor size smaller at diagnosis. The investigators suggested that clinical practice changes, along with greater use of imaging and biochemical studies, were at least partially responsible for the incidence increases. [23]
Race- and age-related demographics
Pheochromocytomas occur in people of all races, although they are diagnosed less frequently in the black population. Pheochromocytomas may occur in persons of any age, but the peak incidence is from the third to the fifth decades of life. Approximately 10% occur in children. Fifty percent of pheochromocytomas in children are solitary intra-adrenal lesions, 25% are present bilaterally, and 25% are extra-adrenal.
A study by Iglesias et al looking at 106 patients with pheochromocytoma found that those diagnosed with the sporadic form of the disease tended to be significantly older than those with familial pheochromocytoma (54.5 years vs 40.8 years, respectively). [24]
Prognosis
The 5-year survival rate for people with nonmalignant pheochromocytomas is greater than 95%. In patients with malignant pheochromocytomas, the 5-year survival rate is less than 50%. [25] Although pheochromocytomas are rare, making the diagnosis is critical because the malignancy rate is 10%, pheochromocytomas may be associated with a familial syndrome, they may precipitate life-threatening hypertension, and the patient may be cured completely with their removal.
A retrospective study by Dhir et al suggested that among patients with pheochromocytoma or paraganglioma, the likelihood of malignancy is greater in persons who are younger, have a larger-sized tumor, or specifically have paraganglioma, as well as in those patients with germline SDHB mutations. Among the patients studied, those with malignancy had a median age of 42 years, versus 50 years for patients without malignancy; the median size of malignant versus nonmalignant tumors was 6.5 cm versus 4 cm, respectively. [26]
Many cardiac manifestations are associated with pheochromocytomas. [27] Hypertension is the most common complication. Cardiac arrhythmias, such as atrial and ventricular fibrillation, may occur because of excessive plasma catecholamine levels. Other complications include the following:
-
Myocarditis
-
Signs and symptoms of myocardial infarction [28]
-
Dilated cardiomyopathy
-
Pulmonary edema: Either of cardiac or noncardiac origin
A pheochromocytoma-induced hypertensive crisis may precipitate hypertensive encephalopathy, which is characterized by altered mental status, focal neurologic signs and symptoms, or seizures. Other neurologic complications include stroke from cerebral infarction or an embolic event secondary to a mural thrombus from dilated cardiomyopathy. Intracerebral hemorrhage also may occur, because of uncontrolled hypertension.
Pheochromocytoma during pregnancy is extremely rare (0.002% of all pregnancies), [29] but undiagnosed pheochromocytoma that occurs during pregnancy carries a grave prognosis, with maternal and fetal mortality rates of 48% and 55%, respectively. However, maternal mortality is virtually eliminated and the fetal mortality rate is reduced to 15% if the diagnosis is made antenatally.
A study by Yokomoto-Umakoshi et al demonstrated evidence for the frequent coexistence of osteoporosis and atherosclerosis in patients with pheochromocytoma. In the presence of vertebral fracture, patients with pheochromocytoma had a 58% prevalence of abdominal aortic calcification (AAC), compared with 6% in pheochromocytoma patients without vertebral fracture. Moreover, a correlation was found between the degree of catecholamine excess and the existence of vertebral fracture and AAC. [30]
-
Axial, T2-weighted magnetic resonance imaging (MRI) scan showing large left suprarenal mass of high signal intensity on a T2-weighted image. The mass is a pheochromocytoma.
-
Abdominal computed tomography (CT) scan demonstrating left suprarenal mass of soft-tissue attenuation representing a paraganglioma.
-
Adrenalectomy specimen containing pheochromocytoma. Non-neoplastic adrenal cortex (yellow) surrounds a small tan-red tumor in the medullary region, representing a pheochromocytoma.
-
H and E, high power, showing classic "balls of cells" feature of a pheochromocytoma. Endocrine tumors such as a pheochromocytoma typically show some degree of nuclear pleomorphism ("endocrine atypia") which does not indicate malignancy.






