Practice Essentials
The term “pernicious anemia” is an anachronism—it dates from the era when treatment had not yet been discovered, and the disease was fatal—but it remains in use to refer to an autoimmune disorder that affects the production of intrinsic factor (IF) by the gastric mucosa. IF binds cobalamin (vitamin B12) and facilitates its transport to the terminal ileum for absorption. [1] Impaired IF production leads to vitamin B12 deficiency and megaloblastic anemia.
Pernicious anemia occurs as a result of autoimmune destruction of parietal cells, which secrete IF, or the development of auto-antibodies targeted against IF itself. [2] Other conditions that can result in impaired IF production include gastrectomy and a rare congenital autosomal recessive disorder that manifests with IF deficiency without gastric atrophy.
Causes of megaloblastic anemia other than impaired IF production include folic acid deficiency, altered pH in the small intestine, and lack of absorption of vitamin B12 complexes in the terminal ileum. Thus, pernicious anemia must be differentiated from other disorders that interfere with the absorption and metabolism of vitamin B12 (see DDx and Workup).
Clinical onset of pernicious anemia usually is insidious and vague. The classic triad of weakness, sore tongue, and paresthesias may be elicited but usually is not the chief symptom complex. Typically, medical attention is sought because of symptoms suggestive of cardiac, renal, genitourinary, gastrointestinal, infectious, mental, or neurologic disorders. Blood studies show anemia with macrocytic cellular indices. See Presentation.
Important goals in the management and care of patients with pernicious anemia include the following:
-
Confirm that the patient has cobalamin deficiency.
-
Initiate treatment with cobalamin. Use higher doses of cobalamin in patients with vitamin B12–associated CNS impairment.
-
Provide concurrent treatment with folic acid and cobalamin in patients who demonstrate evidence for folic acid deficiency but also are being evaluated for pernicious anemia until the latter diagnosis has been ruled out, because although folic acid will restore blood counts, it will not prevent the development of subacute combined system degeneration in patients with pernicious anemia.
-
Monitor response and effectiveness of cobalamin supplementation.
-
Administer adequate quantities of cobalamin for the remainder of the patient's life.
-
Evaluate the patient periodically to rule out gastric carcinoma.
For further discussion, see Treatment and Medication.
Go to Anemia, Iron Deficiency Anemia, and Chronic Anemia for complete information on these topics.
Pathophysiology
Cobalamin is an organometallic substance containing a corrin ring, a centrally located cobalt atom, and various axial ligands (see the image below).
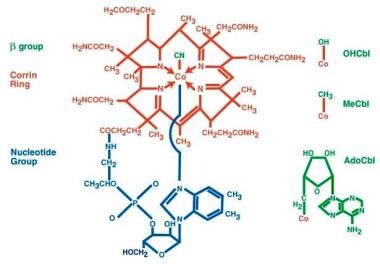 Pernicious anemia. The structure of cyanocobalamin is depicted. The cyanide (Cn) is in green. Other forms of cobalamin (Cbl) include hydroxocobalamin (OHCbl), methylcobalamin (MeCbl), and deoxyadenosylcobalamin (AdoCbl). In these forms, the beta-group is substituted for Cn. The corrin ring with a central cobalt atom is shown in red and the benzimidazole unit in blue. The corrin ring has 4 pyrroles, which bind to the cobalt atom. The fifth substituent is a derivative of dimethylbenzimidazole. The sixth substituent can be Cn, CC3, hydroxycorticosteroid (OH), or deoxyadenosyl. The cobalt atom can be in a +1, +2, or +3 oxidation state. In hydroxocobalamin, it is in the +3 state. The cobalt atom is reduced in a nicotinamide adenine dinucleotide (NADH)–dependent reaction to yield the active coenzyme. It catalyzes 2 types of reactions, which involve either rearrangements (conversion of l methylmalonyl coenzyme A [CoA] to succinyl CoA) or methylation (synthesis of methionine).
Pernicious anemia. The structure of cyanocobalamin is depicted. The cyanide (Cn) is in green. Other forms of cobalamin (Cbl) include hydroxocobalamin (OHCbl), methylcobalamin (MeCbl), and deoxyadenosylcobalamin (AdoCbl). In these forms, the beta-group is substituted for Cn. The corrin ring with a central cobalt atom is shown in red and the benzimidazole unit in blue. The corrin ring has 4 pyrroles, which bind to the cobalt atom. The fifth substituent is a derivative of dimethylbenzimidazole. The sixth substituent can be Cn, CC3, hydroxycorticosteroid (OH), or deoxyadenosyl. The cobalt atom can be in a +1, +2, or +3 oxidation state. In hydroxocobalamin, it is in the +3 state. The cobalt atom is reduced in a nicotinamide adenine dinucleotide (NADH)–dependent reaction to yield the active coenzyme. It catalyzes 2 types of reactions, which involve either rearrangements (conversion of l methylmalonyl coenzyme A [CoA] to succinyl CoA) or methylation (synthesis of methionine).
The basic structure known as vitamin B12 is solely synthesized by microorganisms, but most animals are capable of converting vitamin B12 into the two coenzyme forms, adenosylcobalamin and methylcobalamin. The former is required for conversion of L-methylmalonic acid to succinyl coenzyme A (CoA), and the latter acts as a methyltransferase for conversion of homocysteine to methionine.
When either cobalamin or folate is deficient, thymidine synthase function is impaired. This leads to megaloblastic changes in all rapidly dividing cells because DNA synthesis is diminished. In erythroid precursors, macrocytosis and ineffective erythropoiesis occur.
Severe neurologic impairment, usually subacute combined system degeneration, occurs in cobalamin deficiency. However, vitamin B12 deficiency can also present as peripheral neuropathy, psychosis, or leukoencephalopathy. Neurologic manifestations may occur independently of hematologic manifestations in pernicious anemia. The biochemical impairment in neurologic degeneration may differ from hematologic changes. [3]
Meat and milk are the main dietary sources of cobalamin. Because body stores of cobalamin usually exceed 1000 µg and the daily requirement is about 1 µg, strict adherence to a vegetarian diet for more than 5 years usually is required to produce findings of cobalamin deficiency.
Dietary cobalamin is absorbed in a series of steps, which require proteolytic release from foodstuffs and binding to IF. Subsequently, recognition of the IF-cobalamin complex by specialized ileal receptors—cubilin receptors—must occur for transport into the portal circulation, where it is bound by transcobalamin II (TCII), which serves as the plasma transporter.
The cobalamin-TCII complex binds to cell surfaces and is endocytosed. The transcobalamin is degraded within a lysozyme, and the cobalamin is released into the cytoplasm. An enzyme-mediated reduction of the cobalt occurs with either cytoplasmic methylation to form methylcobalamin or mitochondrial adenosylation to form adenosylcobalamin.
Defects of these steps produce manifestations of cobalamin dysfunction. Most defects become manifest in infancy and early childhood and result in impaired development, intellecual disability, and a macrocytic anemia. Certain defects cause methylmalonic aciduria and homocystinuria. See the image below.
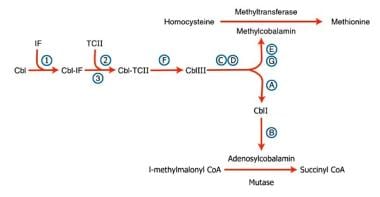 Pernicious anemia. Inherited disorders of cobalamin (Cbl) metabolism are depicted. The numbers and letters correspond to the sites at which abnormalities have been identified, as follows: (1) absence of intrinsic factor (IF); (2) abnormal Cbl intestinal adsorption; and (3) abnormal transcobalamin II (TC II), (a) mitochondrial Cbl reduction (Cbl A), (b) cobalamin adenosyl transferase (Cbl B), (c and d) cytosolic Cbl metabolism (Cbl C and D), (e and g) methyl transferase Cbl utilization (Cbl E and G), and (f) lysosomal Cbl efflux (Cbl F).
Pernicious anemia. Inherited disorders of cobalamin (Cbl) metabolism are depicted. The numbers and letters correspond to the sites at which abnormalities have been identified, as follows: (1) absence of intrinsic factor (IF); (2) abnormal Cbl intestinal adsorption; and (3) abnormal transcobalamin II (TC II), (a) mitochondrial Cbl reduction (Cbl A), (b) cobalamin adenosyl transferase (Cbl B), (c and d) cytosolic Cbl metabolism (Cbl C and D), (e and g) methyl transferase Cbl utilization (Cbl E and G), and (f) lysosomal Cbl efflux (Cbl F).
Intrinsic factor is a gastric protein secreted by parietal cells that is necessary for vitamin B12 absorption. Pernicious anemia is an autoimmune disorder that leads to insufficient intrinsic factor levels either as a result of auto-antibody mediated destruction of parietal cells and/or the intrinsic factor protein itself. Parietal cell auto-antibodies target gastric H+/K+-ATPase. [4] Other disorders that interfere with the absorption and metabolism of vitamin B12 can also result in cobalamin deficiency, with the development of a macrocytic anemia and neurologic complications.
Antiparietal cell antibodies occur in 90% of patients with pernicious anemia but in only 5% of healthy adults. Similarly, binding and blocking antibodies to IF are found in most patients with pernicious anemia. A greater association than anticipated exists between pernicious anemia and other autoimmune diseases, including thyroid disorders, type 1 diabetes mellitus, ulcerative colitis, Addison disease, infertility, and acquired agammaglobulinemia.
An association between pernicious anemia and Helicobacter pylori infections has been postulated but not clearly proven. [5] A higher prevalence of H pylori infection has been reported in patients with pernicious anemia, and eradication of H pylori has had potentially therapeutic effects (eg, reductions in antiparietal cell antibodies, amelioration of atrophic gastritis. However, further research on this hypothesis is warranted. [6]
Cobalamin deficiency may result from dietary insufficiency of vitamin B12; disorders of the stomach, small bowel, and pancreas; certain infections; and abnormalities of transport, metabolism, and utilization (see Etiology). Deficiency may be observed in strict vegetarians. [7] Breastfed infants of vegetarian mothers also are affected. Severely affected infants of vegetarian mothers who do not have overt cobalamin deficiency have been reported. [8]
Classic pernicious anemia produces cobalamin deficiency due to failure of the stomach to secrete IF (see the image below).
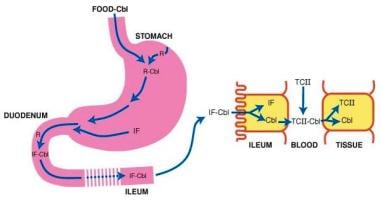 Pernicious anemia. Cobalamin (Cbl) is freed from meat in the acidic milieu of the stomach where it binds R factors in competition with intrinsic factor (IF). Cbl is freed from R factors in the duodenum by proteolytic digestion of the R factors by pancreatic enzymes. The IF-Cbl complex transits to the ileum where it is bound to ileal receptors. The IF-Cbl enters the ileal absorptive cell, and the Cbl is released and enters the plasma. In the plasma, the Cbl is bound to transcobalamin II (TC II), which delivers the complex to nonintestinal cells. In these cells, Cbl is freed from the transport protein.
Pernicious anemia. Cobalamin (Cbl) is freed from meat in the acidic milieu of the stomach where it binds R factors in competition with intrinsic factor (IF). Cbl is freed from R factors in the duodenum by proteolytic digestion of the R factors by pancreatic enzymes. The IF-Cbl complex transits to the ileum where it is bound to ileal receptors. The IF-Cbl enters the ileal absorptive cell, and the Cbl is released and enters the plasma. In the plasma, the Cbl is bound to transcobalamin II (TC II), which delivers the complex to nonintestinal cells. In these cells, Cbl is freed from the transport protein.
In adults, pernicious anemia is associated with severe gastric atrophy and achlorhydria, which are irreversible. The achlorhydria results in a decrease in the release of cobalamin bound to dietary protein. [1] Coexistent iron deficiency is common because achlorhydria prevents solubilization of dietary ferric iron from food. Autoimmune phenomena and thyroid disease frequently are observed. Patients with pernicious anemia have a 2- to 3-fold increased incidence of gastric carcinoma.
Etiology
Cobalamin deficiency may result from the following:
-
Inadequate dietary intake (ie, vegetarian diet)
-
Atrophy or loss of gastric mucosa (eg, pernicious anemia, gastrectomy, ingestion of caustic material, hypochlorhydria, histamine 2 [H2] blockers)
-
Functionally abnormal IF
-
Inadequate proteolysis of dietary cobalamin
-
Insufficient pancreatic protease (eg, chronic pancreatitis, Zollinger-Ellison syndrome [ZES])
-
Bacterial overgrowth in intestine (eg, blind loop, diverticula) – The bacteria compete with the body for cobalamin
-
Diphyllobothrium latum (fish tapeworm) competes with the body for cobalamin
-
Disorders of ileal mucosa (eg, resection, ileitis, sprue, lymphoma, amyloidosis, absent IF-cobalamin receptor, Imerslünd-Grasbeck syndrome, ZES, TCII deficiency, use of certain drugs)
-
Disorders of plasma transport of cobalamin (eg, TCII deficiency, R binder deficiency)
-
Dysfunctional uptake and use of cobalamin by cells (eg, defects in cellular deoxyadenosylcobalamin [AdoCbl] and methylcobalamin [MeCbl] synthesis)
Pernicious anemia is the most common cause of severe vitamin B12 deficiency worldwide and is due to autoimmune destruction of parietal cells and/or intrinsic factor.
Children who develop cobalamin deficiency usually have a hereditary disorder, and the etiology of their cobalamin deficiency is different from the etiology observed in classic pernicious anemia. Congenital pernicious anemia is a hereditary disorder in which an absence of IF occurs without gastric atrophy due to genetic abnormalities that result in failure to secrete IF or production of defective IF. Other gastric conditions that cause cobalamin deficiency are gastrectomy, gastric stapling, and bypass procedures for obesity and extensive infiltrative disease of the gastric mucosa. Usually, these conditions are associated with a decreased ability to mobilize cobalamin from food rather than a malabsorption of cobalamin; thus, such patients may exhibit a normal finding on a Schilling test (stage I).
Pancreatic insufficiency can produce cobalamin deficiency. Nonspecific R binders chelate cobalamin in the stomach, making it unavailable for binding to IF. Pancreatic proteases degrade the R binders and release the cobalamin so that it can bind IF. The cobalamin-IF complex is formed so that it can bind ileal receptors that enable uptake by absorptive cells. Thus, patients with chronic pancreatitis may have impaired absorption of cobalamin.
Cobalamin deficiency is also reported in ZES. The mechanism is believed to be due to the acidic pH of the distal small intestine, which hinders the cobalamin-IF complex from effectively binding to the ileal receptors.
Disorders of the ileum cause cobalamin deficiency as a consequence of the loss of the ileal receptors for the cobalamin-IF complex. Thus, surgical loss of the ileum and diseases such as tropical sprue, regional enteritis, ulcerative colitis, and ileal lymphoma interfere with cobalamin absorption.
Genetic defects of the ileal receptors for IF (ie, Imerslünd-Grasbeck syndrome) and hereditary transcobalamin I (TCI) deficiency produce cobalamin deficiency from birth and are usually discovered early in life.
Many drugs impair cobalamin uptake in the ileum but are rarely a cause of symptomatic vitamin B12 deficiency, because they are not taken for long enough to deplete body stores of cobalamin. Such agents include nitrous oxide, cholestyramine, para -aminosalicylic acid, neomycin, metformin, phenformin, and colchicine.
The clinical manifestations of inherited defects of cobalamin transport and metabolism are usually observed in infancy and childhood. Thus, they are discussed only briefly in this article.
Three hereditary disorders affect absorption and transport of cobalamin, and another seven alter cellular use and coenzyme production. The three disorders of absorption and transport are TCII deficiency, IF deficiency, and IF receptor deficiency. These defects produce developmental delay and a megaloblastic anemia, which can be alleviated with pharmacologic doses of cobalamin. Serum cobalamin values are decreased in the two IF abnormalities but may be within the reference range in TCII deficiency.
The seven abnormalities of cellular use, commonly denoted by letters A through G, can be detected by the presence or absence of methylmalonic aciduria and homocystinuria. The presence of only methylmalonic aciduria indicates a block in conversion of methylmalonic CoA to succinyl CoA and results in either a genetic deficit in the methylmalonyl CoA mutase that catalyzes the reaction or a defect in synthesis of its CoA cobalamin (cobalamin A and cobalamin B deficiency).
The presence of only homocystinuria results either from poor binding of cobalamin to methionine synthase (cobalamin E deficiency) or from producing methylcobalamin from cobalamin and S adenosylmethionine (cobalamin G deficiency). This results in a reduction in methionine synthesis, with pronounced homocystinemia and homocystinuria.
Methylmalonic aciduria and homocystinuria occur when the metabolic defect impairs reduction of cobalamin III to cobalamin II (cobalamin C, cobalamin D, and cobalamin F deficiency). This reaction is essential for formation of both methylmalonic acid and homocystinuria.
Early detection of these rare disorders is important because most patients respond favorably to large doses of cobalamin. However, some of these disorders are less responsive than others, and delayed diagnosis and treatment are less efficacious.
Abnormalities in the intestinal lumen may produce cobalamin deficiency. Individuals with blind intestinal loops, stricture, and large diverticula may develop bacterial overgrowth, which sequesters dietary cobalamin for their metabolic needs. Tapeworm infestation with Diphyllobothrium latum occurs from eating poorly cooked lake fish that are infected and causes cobalamin deficiency because the parasites have a high requirement for cobalamin.
Epidemiology
Adult pernicious anemia usually occurs in people aged 40-70 years. [9] One study found 1.9% of patients older than 60 had undiagnosed pernicious anemia. [10] Congenital pernicious anemia usually manifests in children younger than 2 years.
Whereas the disease originally was believed to be restricted primarily to whites of Scandinavian and Celtic origin, subsequent evidence shows that it occurs in all races. In general, the prevalence of pernicious anemia is probably underestimated, due to the complexity of the diagnosis. [11] A female predominance has been reported in England, Scandinavia, and among persons of African descent (1.5:1). However, data in the United States show an equal sex distribution.
Pernicious anemia likely has a genetic predisposition. The disease is diagnosed more commonly in family members of patients diagnosed with pernicious anemia and is associated with human leukocyte antigen (HLA) types A2, A3, and B7 and blood group type A. Approximately 20% of relatives of patients with pernicious anemia are diagnosed with the same condition,
Patients with pernicious anemia have an increased incidence of autoimmune disorders and thyroid disease, suggesting that the disease has an immunologic component. For example, pernicious anemia may occur together with autoimmune thyroid disease, type 1A diabetes mellitus, alopecia, vitiligo, and chronic atrophic gastritis in type III polyglandular autoimmune (PGA) syndrome—one of a rare group of disorders also known as autoimmune polyendocrine syndromes (APS) and polyglandular failure syndromes. [12]
A population-based cohort study of autoimmune disorders in 22 million individuals in the United Kingdom reported that although in general, the incidence rate of autoimmune disorders was higher in the years 2017-19 compared with 2000-02, the incidence rate of pernicious anemia declined significantly (incidence rate ratio 0.79). [13]
Prognosis
The term pernicious anemia dates from the mid-1800s and reflects the disease's high fatality rate at the time, when its etiology had not yet been discovered. The megaloblastic appearance of cells led many to speculate that it was a neoplastic disease. In the 1920s, however, the response of patients to liver therapy suggested that a nutritional deficiency was responsible for the disorder. [14] This became obvious in clinical trials once vitamin B12 was isolated.
Currently, early recognition and treatment of pernicious anemia provide a normal, and usually uncomplicated, lifespan. Delayed treatment permits progression of the anemia and neurologic complications. If patients are not treated early in the disease, neurologic complications can become permanent. Severe anemia can cause congestive heart failure or precipitate coronary insufficiency.
Although vitamin B12 therapy resolves the anemia, it does not cure the atrophic gastritis, which can progress to gastric cancer. [15] The incidence of gastric adenocarcinoma is 2- to 3-fold greater in patients with pernicious anemia than in the general population of the same age. Presently, periodic gastroscopy and/or barium roentgenographic studies are not advocated in patients with treated pernicious anemia who are asymptomatic, because such screening has not been demonstrated to prolong lifespan.
A population-based, case-control study using the Surveillance, Epidemiology, and End Results (SEER)–Medicare database found that elderly persons with pernicious anemia were not only at significantly increased risk for noncardia gastric adenocarcinoma (odds ratio [OR] 2.18) and gastric carcinoid tumors (OR, 11.43), they were also at increased risk for the following [15] :
-
Tonsillar cancer (OR, 2.00)
-
Hypopharyngeal cancer (OR, 1.92)
-
Esophageal squamous cell carcinoma (OR, 2.12)
-
Small intestinal cancer (OR, 1.63)
-
Liver cancer (OR, 1.49)
-
Myeloma (OR, 1.55)
-
Acute myeloid leukemia (OR, 1.68)
-
Myelodysplastic syndrome (OR, 2.87)
In a longitudinal study of 199 intrinsic factor antibody (IFA)–positive and 168 IFA-negative Chinese patients, Chan et al found that despite a good hematologic response to therapy, both groups had an unsatisfactory neurologic response, and newly diagnosed hypothyroidism was found during follow-up. [16] In addition, newly diagnosed cancers were also found (24 in IFA-positive patients, seven in IFA-negative patients), of which 20% were gastric cancer. [16]
For the IFA-positive patients with a cancer, mean survival was 64 months; for those without a cancer, it was 129 months. Mortality was 31% in this group, in which cancer-related deaths represented 37% of the total. [16] For the IFA-negative patients with a cancer, mean survival was 36 months. For those without a cancer, it was 126 months. Mortality was 21% in this group, in which cancer-related deaths represented 14% of the total.
Chan et al concluded that although Chinese patients treated for pernicious anemia demonstrated a good survival period, they remained at increased risk for gastric carcinoma, and IFA-positive patients had a higher risk of developing all types of cancers and cancer-related deaths than did IFA-negative patients. [16]
Patient Education
Lifelong compliance in obtaining adequate vitamin B12 by injection (or possibly orally) is necessary to avoid relapse of pernicious anemia.
For patient education resources, see Pernicious Anemia (Vitamin B-12 Deficiency) and Vitamin B12.
-
Pernicious anemia. The structure of cyanocobalamin is depicted. The cyanide (Cn) is in green. Other forms of cobalamin (Cbl) include hydroxocobalamin (OHCbl), methylcobalamin (MeCbl), and deoxyadenosylcobalamin (AdoCbl). In these forms, the beta-group is substituted for Cn. The corrin ring with a central cobalt atom is shown in red and the benzimidazole unit in blue. The corrin ring has 4 pyrroles, which bind to the cobalt atom. The fifth substituent is a derivative of dimethylbenzimidazole. The sixth substituent can be Cn, CC3, hydroxycorticosteroid (OH), or deoxyadenosyl. The cobalt atom can be in a +1, +2, or +3 oxidation state. In hydroxocobalamin, it is in the +3 state. The cobalt atom is reduced in a nicotinamide adenine dinucleotide (NADH)–dependent reaction to yield the active coenzyme. It catalyzes 2 types of reactions, which involve either rearrangements (conversion of l methylmalonyl coenzyme A [CoA] to succinyl CoA) or methylation (synthesis of methionine).
-
Pernicious anemia. Inherited disorders of cobalamin (Cbl) metabolism are depicted. The numbers and letters correspond to the sites at which abnormalities have been identified, as follows: (1) absence of intrinsic factor (IF); (2) abnormal Cbl intestinal adsorption; and (3) abnormal transcobalamin II (TC II), (a) mitochondrial Cbl reduction (Cbl A), (b) cobalamin adenosyl transferase (Cbl B), (c and d) cytosolic Cbl metabolism (Cbl C and D), (e and g) methyl transferase Cbl utilization (Cbl E and G), and (f) lysosomal Cbl efflux (Cbl F).
-
Pernicious anemia. Cobalamin (Cbl) is freed from meat in the acidic milieu of the stomach where it binds R factors in competition with intrinsic factor (IF). Cbl is freed from R factors in the duodenum by proteolytic digestion of the R factors by pancreatic enzymes. The IF-Cbl complex transits to the ileum where it is bound to ileal receptors. The IF-Cbl enters the ileal absorptive cell, and the Cbl is released and enters the plasma. In the plasma, the Cbl is bound to transcobalamin II (TC II), which delivers the complex to nonintestinal cells. In these cells, Cbl is freed from the transport protein.
-
Peripheral smear of blood from a patient with pernicious anemia. Macrocytes are observed, and some of the red blood cells show ovalocytosis. A 6-lobed polymorphonuclear leucocyte is present.
-
Bone marrow aspirate from a patient with untreated pernicious anemia. Megaloblastic maturation of erythroid precursors is shown. Two megaloblasts occupy the center of the slide with a megaloblastic normoblast above.
-
Response to therapy with cobalamin (Cbl) in a previously untreated patient with pernicious anemia. A reticulocytosis occurs within 5 days after an injection of 1000 mcg of Cbl and lasts for about 2 weeks. The hemoglobin (Hgb) concentration increases at a slower rate because many of the reticulocytes are abnormal and do not survive as mature erythrocytes. After 1 or 2 weeks, the Hgb concentration increases about 1 g/dL per week.
Tables
Patient Condition |
Methylmalonic Acid |
Homocysteine |
Healthy |
Normal |
Normal |
Vitamin B12 deficiency |
Elevated |
Elevated |
Folate deficiency |
Normal |
Elevated |
Patient Condition |
Stage I No Intrinsic Factor |
Stage II Intrinsic Factor |
Stage III Antibiotic |
Stage IV Pancreatic Extract |
Healthy |
Normal |
… |
… |
… |
Pernicious anemia |
Low |
Normal |
… |
… |
Bacterial overgrowth |
Low |
Low |
Normal |
… |
Pancreatic insufficiency |
Low |
Low |
Low |
Normal |
Defect in ileum |
Low |
Low |
Low |
Low |

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Approach Considerations
- CBC and Peripheral Blood Smear
- Indirect Bilirubin and Serum Lactate Dehydrogenase
- Evaluation of Gastric Secretions
- Serum Cobalamin
- Serum Folic Acid, Methylmalonic Acid, and Homocysteine
- Intrinsic Factor Antibodies
- Schilling Test
- Clinical Trial of Vitamin B12
- Bone Marrow Aspiration and Biopsy
- Other Tests
- Show All
- Treatment
- Medication
- Questions & Answers
- Media Gallery
- Tables
- References






