Practice Essentials
Ovotesticular disorder of sexual development, which was previously termed "intersex," describes disorders in which there is a discrepancy between a person's phenotype, genetic material, and gonads. These discrepancies can be manifested in different gonadal combinations, including ovotestis with ovary, ovary and testis, bilateral ovotestis, and ovotestis and tesis. Ovotestis refers to the histology of a gonad that contains both ovarian follicles and testicular tubular elements. (See the image below.) Such gonads are found exclusively in people with ovotesticular disorder of sexual development (OT-DSD), formerly known as true hermaphroditism. Within the spectrum of DSD, there are varying degrees of discordant genitalia to sex chromosomes. A diagnosis of OT-DSD is based solely on the presence of ovarian and testicular tissue in the gonad and not on the characteristics of the internal and external genitalia, even if ambiguous. [1]
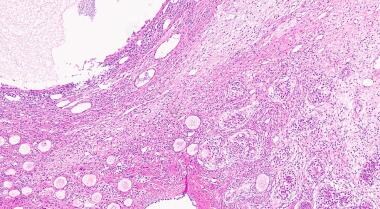 H&E stain of ovotestis demonstrating ovarian parenchyma (left) with multiple primary follicles and testicular parenchyma (right). Courtesy of Cureus [De Jesus Escano MR, Mejia Sang ME, Reyes-Mugica M, Colaco M, Fox J. Ovotesticular Disorder of Sex Development: Approach and Management of an Index Case in the Dominican Republic. Case Report. Cureus. Published 5 Oct 2021. Online at https://www.cureus.com/articles/70052-ovotesticular-disorder-of-sex-development-approach-and-management-of-an-index-case-in-the-dominican-republic].
H&E stain of ovotestis demonstrating ovarian parenchyma (left) with multiple primary follicles and testicular parenchyma (right). Courtesy of Cureus [De Jesus Escano MR, Mejia Sang ME, Reyes-Mugica M, Colaco M, Fox J. Ovotesticular Disorder of Sex Development: Approach and Management of an Index Case in the Dominican Republic. Case Report. Cureus. Published 5 Oct 2021. Online at https://www.cureus.com/articles/70052-ovotesticular-disorder-of-sex-development-approach-and-management-of-an-index-case-in-the-dominican-republic].
Pathophysiology
Patients with ovotesticular disorder of sexual development are individuals who have both ovarian and testicular tissue. This diagnostic nomenclature is applied regardless of the patient's karyotype. The gonads present may be bilateral ovotestes, or they may be a combination of a unilateral ovary or testis with an ovotestis on the contralateral side. Ovotestes are usually compartmentalized, with connective tissue separating the ovarian components from the testicular components. However, on rare occasions, an intermixture of these elements may occur. Additionally, testicular and ovarian tissue may develop on the same side of the pelvis as a separate ovary and testis.
Ovotestes are the most frequent gonad present (60%), followed by the ovary and then the testis (9%). The ovotestis can be anatomically located in an ovarian position, or along the anatomical path of the testes in the labioscrotal fold, the inguinal canal, or at the internal inguinal ring. Ovaries, when found, can occupy the normal abdominal position, although they may occasionally be found at the internal inguinal ring. Interestingly, ovaries occur more commonly on the left side than the right. The reason for this predilection is unknown. Testes are usually found in the scrotum, although they can be found at any level along the path of embryonic descent from abdomen to scrotum, frequently presenting as inguinal hernias.
Ovaries and ovarian portions of ovotestes appear normal and demonstrate follicular growth with estradiol production. Approximately 50% of ovotestes show evidence of ovulation. The presence of estradiol in developing ovarian follicles usually inhibits spermatogonia development in adjacent or contralateral seminiferous tubules. Degeneration and hyalinization of the seminiferous tubules with poor germ cell development is frequently observed. In all documented biopsied cases, there is a significant decline in germ cell development and an increase in tubular sclerosis by puberty. Leydig cell hyperplasia may also occur with aging. Spermatogenesis in testis and ovotestis is rare given the high circulating estrogen levels.
Internal duct development usually corresponds to the adjacent gonad. Many patients with ovotesticular disorder of sexual development have a uterus. Müllerian duct structures typically develop on the gonadal side not containing testicular tissue. Wolffian duct structures tend to be observed on the gonadal side containing functioning testicular tissue.
People with OT-DSD are born with ambiguous genitalia. Historically, most affected individuals have been reared as males due to the size of the phallus. Most have varying degrees of labioscrotal fusion and/or hypospadias. However, because of functioning normal ovarian tissue, most people experience breast development at puberty, and approximately two-thirds of those with a 46,XX peripheral karyotype menstruate. [2] A report describes the rare case of a patient with nearly normal-appearing external male genitalia, in whom the diagnosis was not suspected until progressive bilateral breast enlargement occurred during adolescence. [3]
Etiology
Normal sexual differentiation is based on genetic sex (XX or XY), which is established at conception. Until 7 weeks of gestation, the fetus is sexually indifferent, internally developing both wolffian and mullerian ducts. Expression of sex-determining genes on the early bipotential gonad promotes development of the testis or ovary.
Various genes expressed by the Y chromosome at very specific times during development are responsible for the differentiation of the testes. A 35-kilobase (kb) gene determinant located on the distal short arm of the Y chromosome, known as the SRY (sex determining region of the Y chromosome) is responsible for initiating testes formation. SRY codes for a transcription factor that acts in the somatic cells of the genital ridge. The transient expression of this gene triggers a cascade of events that leads to the development of testicular Sertoli and Leydig cells. SRY expression directs testicular morphogenesis, characterized by the production of MIS (müllerian-inhibiting substance), and, later, testosterone.
Surprisingly, more than half of the patients with XX ovotesticular disorder of sexual development lack SRY, despite the presence of testicular differentiation. This suggests that this gene codes for a product that reacts with other genes on Y, X, and/or autosomes to complete testicular differentiation. Research has looked at SRY-related high mobility box 9 (SOX9) gene, located on autosomal chromosome 17, as a contributor to Sertoli cell differentiation. Increased expression of SOX9 is being studied as a cause for female-to-male sex reversal in 46,XX SRY negative people with ovotesticular disorder of sexual development. Recently, an inactivating mutation of SOX9 was shown to be associated with autosomal sex reversal and camptomelic dysplasia.
The DAZ (deleted in azoospermia) gene family consists of a cluster of genes on the Y chromosome that give rise to proteins that influence male germ cell differentiation. In humans, deletion of any 1 of 3 DAZ regions (ie, AZFa, AZFb, AZFc) disrupts spermatogenesis. Today, deletion of the AZFc region of the Y chromosome is the most frequent molecularly defined cause of spermatogenic failure.
In 46,XY males, the Sertoli cells of the testes are responsible for the production of mullerian-inhibiting substance, which causes regression of the mullerian ducts. The Leydig cells then produce testosterone, which promotes the development of the epididymis, vas deferens, and seminal vesicles.
For the fetus exposed to only the X chromosome, female gonadal development ensues. Ovarian differentiation appears to rely on a mechanism that is triggered mostly, but not solely by the absence of the testicular determinant. Female development is no longer viewed as only a default pathway for reproductive differentiation. In humans, a complete 46,XX chromosomal complement is necessary for normal ovarian differentiation. Autosomal genes also appear to be involved in ovarian maintenance. Properties of the X-linked gene DAX1 (Dose sensitive sex reversal locus on X chromosome, gene 1) suggest that this gene is important in ovarian determination. Investigators have postulated that the DAX1 gene product may actually be an anti-testes factor and may be antagonistic to the action of SRY. [4] An additional signaling molecule, Wnt4, is found in mullerian ducts and contributes to the development of female internal genitalia.
Internal genitalia of the female fetus develop if there is no exposure to the SRY gene and its signaling molecules. The wolffian duct regresses and the mullerian duct then matures into the oviduct, uterus, cervix, and upper vagina.
Hormone expression during the 9th week of gestation, from the testes or ovary, stimulates external genitalia development. By the 14th week of gestation the external genitalia have been formed. During the developmental process, there are multiple opportunities for errors in differentiation, all of which have been theorized as possible causes of the ovotesticular disorder of sexual development.
In humans, genetic sex has traditionally been evaluated through establishing the karyotype of peripheral lymphocytes. However, the peripheral karyotypes of patients with OT-DSD show marked variation. Approximately 60% are 46,XX; 15% are 46,XY; and 25% show various forms of mosaicism. Less than 1% show 46,XX/46,XY chimerism or the existence of 2 or more cell lines, each of which has a different genetic origin.
Therefore, ovotesticular disorder of sexual development is a genetically heterogeneous condition. Phenotypic, gonadal, and molecular studies have led to several causation theories:
-
Genetic chimerism: Fewer than 1% of people with OT-DSD have 46,XX/46,XY chimerism or the existence of 2 or more cell lines, each of which has a different genetic origin. Chimerism can result from several events.
Dispermic chimerism (double fertilization) can arise from fertilization of the secondary oocyte and first polar body, fertilization of the ovum and the first polar body, or fertilization of the ovum and the second polar body.
Chimerism can also arise as an exchange of cells between dizygous twins of different sex (ie, fusion of 2 embryos).
-
Nondisjunction: Postzygotic mitotic errors arising from anaphase lag may occur in 45,X/46,XY or 45,X/46,XY/47,XYY mosaicism. Note, however, that most 45,X/46,XY individuals have mixed gonadal dysgenesis as opposed to true hermaphroditism.
-
X-Y translocation: Paternal meiotic exchange between the pseudoautosomal regions of chromosomes X and Y could provide a mechanism for the translocation Y-chromosomal sequences, including SRY onto an X chromosome in some forms of 46,XX testicular differentiation.
-
Mutation: A mutation of a gene on the X chromosome or alternatively on an autosome that allows testis determination without the SRY gene could explain some forms of 46,XX testicular differentiation. In addition, some 46,XX with OT-DSD have been observed to have a translocation of SRY onto the X chromosome. However, most individuals with ovotesticular disorder of sexual development with 46,XX are SRY negative.
-
Occult mosaicism: Although most people with OT-DSD have a 46,XX peripheral karyotype, recent case reports have documented the detection of occult mosaicism in the gonads of some of these individuals through molecular techniques. Polymerase chain reaction (PCR) has identified SRY -positive tissue in gonads from several, but not all, people with 46,XX ovotesticular disorder of sexual development.
Mutation of downstream autosomal genes involved with testicular differentiation and mutation/duplication or deletion of an X-linked locus may explain SRY –negative ovotesticular disorder of sexual development.
Epidemiology
International statistics
Ovotesticular disorder of sexual development is a rare condition. Most cases have a sporadic distribution, although there are a few documented cases of familial recurrence. Genital ambiguity occurs in 1 in 4500 births, and ovotesticular disorder of sexual development occurs in fewer than 10% of all disorders of sexual development. More than 400 cases have been reported worldwide. [5]
Race- and age-related demographics
Geographic variation has been noted, with the highest incidence occurring in the Black population of southern Africa.
Despite the fact that most people with true hermaphroditism present with genital ambiguity, less than 20% are diagnosed before age 5 years. Seventy-five percent are diagnosed by age 20 years.
Prognosis
Aside from the physical and emotional consequences associated with genital ambiguity, patients with ovotesticular disorder of sexual development usually do not possess other developmental malformations.
These individuals generally are of average intelligence and have a normal life expectancy.
Fertility potential does exist in people with ovotesticular disorder of sexual development who are given a female sexual assignment. Ovulation can occur, and several pregnancies have been reported in this group. To date, all documented offspring have been male. One report describes a 46,XX/46,XY infertile chimeric male who fathered a child with sperm obtained from his testicular tissue through intracytoplasmic sperm injection. [6, 7, 8]
Another case report describes the successful use of in vitro fertilization in a patient with a normal uterus and left gonad who underwent unilateral gonadectomy as a child. She delivered a healthy male infant. [9]
People with ovotesticular disorder of sexual development who are given a male sex assignment rarely reproduce. Spermatogenesis has been reported in only 12% of these cases, and tubular atrophy with hypoplastic testicular tissue is the norm. There are only 3 reported cases of males with OT-DSD fathering children.
Patients should be counseled that infertility is common despite excision of discordant tissue and genital reconstruction.
Many patients with ovotesticular disorder of sexual development are sexually active with a small portion being sexually dissatisfied. Those raised as males may complain of an inability to have and/or maintain erections, while females may complain of vaginal stenosis, recurrent cystitis, and hot flushes. [10]
Morbidity/mortality
Neoplasia
-
Gonadal tumors with malignant potential occur in 2.6% of all cases of ovotesticular disorder of sexual development. The testis or testicular component of an ovotestis is likely to be dysgenetic; dysgerminomas, seminomas, gonadoblastomas, and yolk sac carcinomas have all been reported.
-
Those with the 46,XY karyotype are at the greatest risk of developing a gonadal malignancy. Benign tumors, including mucinous cystadenomas, benign teratomas, and Brenner tumors, have also been reported.
-
If a testis is located in the scrotum, maintaining rigorous follow-up with sonography and/or pelvic MRI is prudent, and a biopsy after puberty is indicated to detect early premalignant or malignant transformation.
-
One case report of a 47-year-old 46,XX/46,XY woman with a malignant phyllodes tumor in the right breast and an invasive lobular carcinoma in the left breast suggests a modified breast cancer risk similar to that observed in Klinefelter syndrome. [11]
-
Another case reported an invasive squamous cell carcinoma of the vagina, serving as a reminder that malignant changes can occur in residual müllerian tissue. [12]
Obstructed genital tract
Cryptomenorrhea, hematometra, and lower abdominal pain associated with endometriosis may occur in individuals with cervical atresia or other forms of müllerian duct anomalies.
Hernias and cryptorchism
Because of malposition of the gonads, gonadal torsion, and associated duct structures, a variety of organs have been encountered within the inguinal canal, and inguinal hernias are a common occurrence. Complications associated with undescended or partial testicular descent also may be encountered.
Complications
Traditionally, complications from the surgical treatment of patients with disorders of sexual development were related to stenosis after vaginal reconstruction procedures and urinary tract problems associated with correction of severe hypospadias.
Today, patients are diagnosed and operated on earlier and tend to have fewer complications.
Gender assignment should be made prior to age 18 months, when children develop gender identity, so as to minimize psychosocial trauma.
Patient Education
Once the diagnosis of genital ambiguity is made, ongoing psychological support for the patient, parents, and other family members is critical. [13]
Psychological counseling is perhaps even more important for patients with a diagnosis delayed until puberty or adult life.
-
MRI demonstrating uterus and vagina in a 46XY infant. Courtesy of BMC Springer Nature [Scarpa MG, Lesma A, Di Grazia M, et al. Ovotesticular differences of sex development: male or female? Case series. Italian Journal of Pediatrics. 30 May 2019; 45;66. Online at https://ijponline.biomedcentral.com/articles/10.1186/s13052-019-0660-8].
-
Intraoperative finding of uterus, fallopian tube, and streak gonad in 46XY infant. Courtesy of BMC Springer Nature [Scarpa MG, Lesma A, Di Grazia M, et al. Ovotesticular differences of sex development: male or female? Case series. Italian Journal of Pediatrics. 30 May 2019; 45;66. Online at https://ijponline.biomedcentral.com/articles/10.1186/s13052-019-0660-8].
-
H&E stain of ovotestis demonstrating ovarian parenchyma (left) with multiple primary follicles and testicular parenchyma (right). Courtesy of Cureus [De Jesus Escano MR, Mejia Sang ME, Reyes-Mugica M, Colaco M, Fox J. Ovotesticular Disorder of Sex Development: Approach and Management of an Index Case in the Dominican Republic. Case Report. Cureus. Published 5 Oct 2021. Online at https://www.cureus.com/articles/70052-ovotesticular-disorder-of-sex-development-approach-and-management-of-an-index-case-in-the-dominican-republic].






