Practice Essentials
Nephrocalcinosis is a condition in which calcium levels in the kidneys are increased. Most often, the increase in renal calcium is generalized, as opposed to the localized increase observed in calcified renal infarct and caseating granulomas of renal tuberculosis. See the image below.
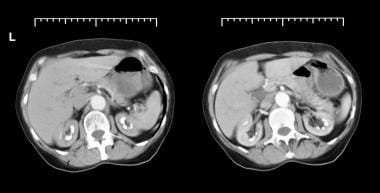 Axial CT scans from patient with long history of renal tubular acidosis. Images show bilateral medullary nephrocalcinosis (early arterial phase).
Axial CT scans from patient with long history of renal tubular acidosis. Images show bilateral medullary nephrocalcinosis (early arterial phase).
Signs and symptoms
Presentation is primarily determined by the underlying etiology, though in many cases the condition remains asymptomatic and is identified only as a radiologic abnormality. The physical findings are nonspecific and reflect the underlying disorders responsible.
Clinical features of hypercalcemic nephropathy may include the following:
-
Relative vasopressin resistance with decreased renal concentrating ability and increased free water diuresis, manifesting as polyuria and polydipsia
-
Renal glycosuria, reduced glucose tubular maximum, aminoaciduria, and nonglomerular proteinuria
-
Reversible hypertension
-
Kidney failure, usually reversible but sometimes not
Clinical features of microscopic nephrocalcinosis (on the basis of animal studies) may include the following:
-
Reduced concentration capacity
-
Increased blood urea nitrogen (BUN)
-
Prolongation of nephron transit time in the distal tubule
-
Acute pyelonephritis or calculous ureteral obstruction with kidney failure
Clinical features of macroscopic nephrocalcinosis (the form most commonly seen) may include the following:
-
Renal colic
-
Hematuria
-
Passage of urinary stones
-
Urinary tract infection
-
Polyuria and polydipsia
-
Hypertension
-
Proteinuria
-
In Dent disease, loss of low-molecular-weight proteins, hypercalciuria, and nephrolithiasis
-
Microscopic pyuria
-
Distal tubular dysfunction with a mild salt-losing defect
-
Proximal tubular dysfunction (unusual)
-
Secondary distal tubular acidosis
-
Kidney failure
See Presentation for more detail.
Diagnosis
Laboratory studies that may be useful include the following:
-
Serum calcium, phosphate, and albumin levels
-
Blood urea nitrogen (BUN) and serum creatinine levels
-
Estimated glomerular filtration rate (eGFR)
-
Serum potassium concentration
-
Urinalysis and urine culture
-
Assessment of 24-hour urinary excretion of calcium, oxalate, citrate, and uric acid
-
Urinary magnesium levels
-
Parathyroid hormone and thyroid-stimulating hormone levels
Imaging studies that may be considered include the following:
-
Radiography (eg, kidney-ureter-bladder [KUB])
-
Ultrasonography (more sensitive than conventional radiography)
-
Computed tomography (CT; more effective in detecting calcification)
Magnetic resonance imaging (MRI) offers no advantages over these modalities and is not warranted unless another compelling indication is present.
See Workup for more detail.
Management
Pharmacologic and other nonsurgical treatments for hypercalcemia and hypercalcemic nephropathy include the following:
-
Adequate hydration with an isotonic sodium chloride solution (the single most effective measure for reversing hypercalcemia and protecting the kidneys)
-
Cinacalcet (for correction of hyperparathyroidism)
-
Chemotherapeutic agents (for osteolytic malignancies)
-
Steroids (to decrease intestinal calcium absorption and vitamin-D activity)
-
Hydroxychloroquine (for sarcoid granulomas)
-
Calcitonin or bisphosphonates (to inhibit bone resorption)
Pharmacologic and other nonsurgical treatments for macroscopic nephrocalcinosis include the following:
-
Thiazide diuretics (eg, hydrochlorothiazide)
-
Dietary salt restriction
-
Potassium and magnesium supplementation
-
Citrate supplementation (preferably as potassium citrate)
-
High-dose pyridoxine
Surgery options that may be considered for urinary stones causing obstruction include the following:
-
Percutaneous nephrolithotomy
-
Laser and shock wave lithotripsy
-
Stent placement
-
Open surgery (rarely necessary)
Parathyroidectomy may be considered for removal of enlarged adenomas.
See Treatment and Medication for more detail.
Background
Nephrocalcinosis is a condition in which calcium levels in the kidneys are increased. There is predominantly interstitial deposit of calcium phosphate or calcium oxalate in the renal cortex and/or medulla. This condition can sometimes overlap with nephrolithiasis, which is characterized by intratubular deposits of calcium. Calcium deposits can be detected (usually as an incidental finding) through a radiologic examination or via microscopic examination of the renal tissues. The term nephrocalcinosis most often applies to a generalized increase in interstitial renal calcium content, as opposed to the localized increase observed in calcified renal infarct and caseating granulomas of renal tuberculosis. [1]
Nephrocalcinosis has a substantial overlap with hypercalcemia, nephrolithiasis, renal parenchymal damage, and reduced renal function. Therefore, rather being considered a single, distinct disease process, it should be viewed as a helpful finding for several distinct disease processes, one that demands evaluation. [1]
Microscopic nephrocalcinosis is characterized by the presence of microscopic crystalline calcium precipitates in the form of oxalate or phosphate. Patients with macroscopic nephrocalcinosis have larger areas of calcifications, which can be observed on visual or radiologic examination without further magnification.
Pathophysiology
Hypercalcemic nephropathy (chemical nephrocalcinosis)
Patients with hypercalcemia develop renal function abnormalities. Under these circumstances, the term hypercalcemic nephropathy is more appropriate than is the older term chemical nephrocalcinosis.
Calcium is a critical divalent cation that is transported, along with sodium, potassium, and water, in a complex and regulated manner along the renal tubular epithelium. The cytoplasmic concentration of calcium is tightly regulated and kept very low, being maintained by active extracellular extrusion of calcium and sequestration into the endoplasmic reticulum and mitochondria. Increased extracellular calcium leads to impairment of the calcium messenger system with gross tubular impairment.
The effects of increased calcium have been studied extensively in rats. Rats treated with vitamin D demonstrated mitochondrial swelling and loss of mitochondrial enzyme activities before calcification appeared. Parathyroid extract−induced hypercalcemia was found to cause changes in rat kidneys, predominantly affecting the distal nephron, with focal necrosis of the outer medullary collecting ducts and the ascending limb of the loop of Henle.
Hypercalcemia results in renal vasoconstriction and a reduced glomerular filtration rate. It also interferes with renal tubular functions. Impaired renal concentration ability and resistance to vasopressin are the most common defects observed with hypercalcemia. These changes may be mediated by reduced sodium transport in the loop of Henle (see the image below) and by antidiuretic hormone antagonism via calcium-sensing receptors, [2] or they may be related to medullary prostaglandin synthesis.
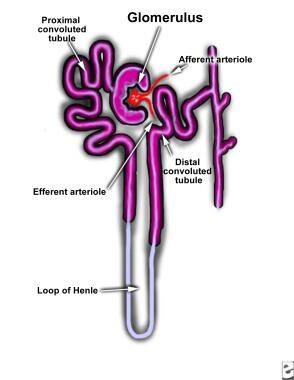
Maximum diluting capacity remains unimpaired. Effectively, the sum effect of this process will be a clinical picture resembling that of nephrogenic diabetes insipidus.
Renal sodium conservation is also impaired because of reduced absorption of sodium chloride in the medullary thick ascending limb and collecting tubule, although this rarely results in gross renal sodium losses. Potassium excretion is increased. Magnesium excretion is also increased, probably due to suppression of parathyroid hormone, which enhances tubular magnesium absorption. [3]
Hypercalcemia increases urinary calcium excretion by increasing the filtered load and reducing tubular absorption. Its effects on phosphate excretion are complex. In experimental animals, pure hypercalcemia reduces phosphate excretion; conversely, in certain cancers, it can be associated with increased phosphate excretion, but the latter occurrence is probably due to the presence of phosphaturic peptides (phosphatonins), which are secreted in some malignancies. [4, 5]
The effects on the acid-base balance are even more complex. Increased renal acid excretion occurs with intravenous (IV) calcium infusions, and metabolic alkalosis has frequently been reported in patients with hypercalcemia. On the other hand, parathyroid hormone decreases hydrogen ion excretion, leading to a distal type of renal tubular acidosis (RTA).
This opposing effect of hypercalcemia and parathyroid hormone has been used in the differential diagnosis of hypercalcemia, because serum bicarbonate is lower and chloride is higher when hyperparathyroidism is the cause of hypercalcemia.
Microscopic nephrocalcinosis
Microscopic nephrocalcinosis has undergone considerable laboratory study. Although this condition theoretically occupies a stage between hypercalcemia and macroscopic nephrocalcinosis, it is difficult to demonstrate in humans, because renal biopsies are not routinely performed in the early stages of metabolic diseases known to lead to the macroscopic stage. However, some elegant human data are now available that demonstrate early stone formation, with blockage of the collecting tubes and subsequent inflammatory response. [6]
At autopsy, healthy human kidneys invariably contain microscopic deposits of calcium in the renal medulla. Microscopic nephrocalcinosis can occur without macroscopic involvement in patients with longstanding hypercalcemia from primary parathyroidism, milk-alkali syndrome, or primary hyperoxaluria.
Different patterns of microscopic nephrocalcinosis have been described. The cortical pattern has been found after parenteral calcium administration. The corticomedullary type involves calcium phosphate deposits that occur in the inner zone of the renal cortex and extend into the medulla. Precipitating factors include excess parathyroid hormone, vitamin D, acetazolamide, magnesium depletion, decreased urinary citrate, and hypothyroid state. Increased plasma calcium is not an essential prerequisite for this type of nephrocalcinosis.
The medullary pattern has been reported in hyaline droplet nephropathy resulting from inhalation of volatile hydrocarbons. The pelvic type affects renal papillae. The deposits usually are calcium phosphate, but calcium oxalate also has been implicated. The underlying mechanism appears to be either increased intestinal absorption or decreased renal excretion of calcium.
Macroscopic nephrocalcinosis
Macroscopic nephrocalcinosis refers to calcium deposition that is visible without magnification and usually is discovered by means of conventional radiography, ultrasonography, or computed tomography (CT) or at autopsy. Macroscopic nephrocalcinosis can affect either the cortex or the medulla, with the latter site being more commonly involved.
Cortical nephrocalcinosis is rare and usually occurs secondary to diffuse cortical disease injury. The calcification can be patchy or confluent. In chronic glomerulonephritis, calcium deposits are found most often in periglomerular tissue and not in the glomerulus. Nephrocalcinosis also has been reported in familial infantile nephrotic syndrome and in Alport syndrome.
Acute cortical necrosis secondary to toxemia of pregnancy, snakebite, or hemolytic-uremic syndrome can lead to patchy cortical nephrocalcinosis. Calcium deposition can start as early as 30 days after cortical necrosis. Chronic pyelonephritis and vesicoureteral reflux are also implicated. [7] Kidney transplantation, primary hyperoxaluria, methoxyflurane abuse, autosomal recessive polycystic kidney disease, and benign nodular cortical nephrocalcinosis may be involved in cortical nephrocalcinosis, albeit rarely.
Medullary nephrocalcinosis assumes the form of small nodules of calcification clustered in each pyramid. Diagnosing the underlying renal disease on the basis of the appearance is difficult. Characteristic exceptions include papillary necrosis due to analgesic abuse and medullary sponge kidneys. [8] In papillary necrosis, the entire papilla may be calcified, whereas in medullary sponge kidney, there is a characteristic band of calcification in the renal pyramids.
It has been suggested that when hypercalcemia is the most important factor, the first foci of calcification develop in the renal tubular cells, whereas when hypercalciuria is the major factor, the initial foci form in the interstitium.
Intraluminal tubular calcium crystals are believed to serve as potential nidi for further buildup of calcium and other stone-forming substances, including oxalate and uric acid. Whether further growth of nephroliths occurs probably depends on a number of additional factors, such as abnormal urine composition, urine flow and volume, and the presence or absence of endogenous inhibitors of crystalline formation in the urine.
Etiology
Etiologies of cortical calcium deposition in kidneys are as follows:
-
Pyelonephritis
-
Tuberculosis
-
Glomerulonephritis
-
Acute cortical deposits
-
Acute transplant rejection
-
Alport syndrome
Etiologies of medullary calcium deposition in kidneys are as follows:
-
Hyperparathyroidism
-
Distal renal tubular acidosis (RTA)
-
Vitamin D overdose
-
Primary hyperoxaluria
-
Medullary sponge kidney
-
Bartter syndrome
-
ADCK4 mutations
Risk factors for nephrocalcinosis include the following:
-
Persistent hypercalcemia and hyperphosphatemia.
-
Increased urinary excretion of calcium, phosphate, or oxalate
-
Hypocitraturia
-
Chronic hypokalemic states
-
Hyperoxaluria
Primary hyperparathyroidism is the single most common cause of nephrocalcinosis in adults. Although nephrocalcinosis is a relatively rare complication (5%), primary hyperparathyroidism itself is relatively common, especially in the elderly.
Nephrocalcinosis is related more to the duration than to the intensity of hypercalcemia. The classic clinical findings are sometimes referred to as “(abdominal) groans, stones, and bones.” This common phrase is a reminder that patients may present with kidney stones, bone pain, osteoporosis, and pathologic fractures, all of which can result in abdominal discomfort. Rarely, hyperparathyroidism can be associated with multiple endocrine neoplasia type 1 (MEN1).
Patients with chronic hypoparathyroidism are at an increased risk for nephrocalcinosis. [9]
Distal RTA is the second most common cause of medullary nephrocalcinosis. Both the familial form and the secondary form (autoimmune-associated anti-K/H channel antibody) are common. [10] The mechanisms contributing to nephrocalcinosis in distal RTA are hypercalcemia, hypercalciuria, metabolic acidosis, and reduced citrate excretion in the presence of increased urinary pH. [11] Because medullary nephrocalcinosis itself can cause distal RTA, distinguishing the initial insult can be difficult. Kidney function is fairly well maintained.
Other causes of nephrocalcinosis are hypervitaminosis-D states [12] resulting from excessive treatment of hypoparathyroidism, self-administration of vitamins, and the presence of a granulomatous disease, such as sarcoidosis. [13] Nephrocalcinosis can be among the renal manifestations of primary mitochondrial syndromes (eg, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes [MELAS] syndrome; Kearns-Sayre syndrome; Leigh syndrome; mitochondrial depletion syndromes). [14]
In granulomatous disorders, conversion of 25-hydroxycholecalciferol to 1,25-dihydroxycholecalciferol in the granuloma is increased, resulting in an unregulated production of bioactive vitamin D with resultant excessive intestinal absorption of calcium and phosphorus. In addition, cytokines (eg, interleukin [IL]-2) released in these disorders cause dysregulation of calcium homeostasis and activation of osteoclasts, resulting in subacute and chronic hypercalcemia.
Any other cause of hypercalcemia, particularly when associated with hypercalciuria, can contribute to nephrocalcinosis. Etiologies include milk-alkali syndrome (due to excess ingestion of antacids), hyperparathyroidism, and malignant disease (due to bone involvement and humoral factors, including cytokines and parathyroid hormone-related peptide). Idiopathic hypercalciuria, [15, 16] a common metabolic disease, is also known to cause nephrocalcinosis.
Nephrocalcinosis and kidney failure are increasingly recognized as common complications of phosphate supplementation, particularly in the elderly. [17, 18, 19, 20, 21] Other possible risk factors are preexisting kidney failure, high blood pressure, and pharmacologic treatment of high blood pressure (eg, with angiotensin-converting enzyme [ACE] inhibitors or angiotensin-receptor blockers [ARBs]).
Phosphate supplements may contribute to kidney calcifications in children with hypophosphatemic rickets. In vitro studies have shown that an increased urinary concentration of phosphate can result in intratubular crystallization with altered solubility.
Medullary sponge kidney is a common cause of medullary calcification, with calcium lying in dilated collecting ducts rather than in the renal substance. These ectatic outpouchings are believed to be areas of urinary stasis possessing the ideal characteristics for fostering the formation of these calcifying complexes. The calcium deposits are larger and more sharply defined than they are in metabolic disease, and they are uneven in distribution. [8] Associated hemihypertrophy of the body may exist.
Unlike the severe kidney damage with minimal calcification associated with hypercalcemic states, nephrocalcinosis associated with distal RTA and medullary sponge kidney usually is gross, and kidney function is relatively well preserved.
Nephrocalcinosis has been reported in patients receiving antiepileptic drug therapy with topiramate. [22] Classic analgesic nephropathy, which sometimes manifested as nephrocalcinosis and was characterized pathologically by renal papillary necrosis, has disappeared since the removal of phenacetin from the market internationally. [23]
Other associations with nephrocalcinosis include rapidly progressive osteoporosis due to immobilization, menopause, aging, or steroids.
Medullary calcification can be induced by primary (familial) hyperoxaluria or by secondary hyperoxaluria due to increased intake of oxalates, enhanced absorption resulting from intestinal disease, or ingestion of ethylene glycol or methoxyflurane. [24, 25] Primary hyperoxaluria and ethylene glycol intoxication are also associated with diffuse calcium-oxalate depositions in many other organs, including the eye and the heart.
Chronic hypokalemic states, such as Bartter syndrome, primary hyperaldosteronism, Liddle syndrome, and 11-beta hydroxylase deficiency, are associated with reduced urine citrate excretion and tubular epithelial damage, leading to calcium precipitations. Nephrocalcinosis is most frequently associated with Bartter I, II and V. [26]
Mutations in ADCK4, one of the genes causing steroid-resistant nephrotic syndrome, usually manifest as focal segmental glomerulosclerosis with onset in late childhood. ADCK4 interacts with the coenzyme Q10 (CoQ10) biosynthesis pathway, and in its early stage this condition is treatable with CoQ10 supplementation. [27]
Autosomal dominant hypophosphatemic rickets and X-linked hypophosphatemic conditions [28] have been associated with abnormal phosphate wastage and nephrocalcinosis due to elevated levels of phosphatonins (fibroblast growth factor 23; secreted frizzled-related protein 4). [4, 5] Nephrocalcinosis is very common (frequency ~80% on ultrasonography) and may be associated with phosphate supplementation for the condition.
Dent disease and familial magnesium-losing nephropathy are rare inherited diseases causing medullary calcification.
Dent disease (also referred to as X-linked recessive hypophosphatemic rickets [in Italy], X-linked recessive nephrolithiasis, and idiopathic low-molecular weight proteinuria with hypercalciuria and nephrocalcinosis [in Japan]), arises from a defect in a gene on the short arm of the X chromosome that codes for the renal chloride channel in the proximal tubule (CLC-5). Mutations in the OCRL-1 gene—normally associated with Lowe syndrome—have been described in cases of clinical Dent disease, expanding the potential for genetic diversity. [29]
Inherited forms of magnesium-losing nephropathy have been described. [30] Familial hypomagnesemia hypercalciuric nephrocalcinosis (FHHNC) is an autosomal recessive disease associated with cation loss through a defect in a renal tight junction protein claudin-16 (paracellin-1) [31, 32, 33] or claudin-19 [34, 35] involved in paracellular transport. Claudin-16/paracellin-1 is dysfunctional in familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC). However, hypomagnesemia is not a mandatory finding in all kindreds. [36] Thiazide-type diuretics appears efffective in lessning hypercelcuria and nephrocalcinosis.
Associated malignancies are not typical in nephrocalcinosis, because patients seldom survive long enough with hypercalcemia to develop them; a possible exception is parathyroid carcinoma.
Familial benign hypercalcemia and hyperthyroidism are not associated with kidney calcification.
Premature sick infants have been observed to develop diffuse nephrocalcinosis (noted in about two thirds of infants with birth weights of < 1500 g), typically when exposed to diuretic therapy or prolonged oxygen therapy. The natural history of this phenomenon is not well understood, and no clearly effective treatment has been established. [37, 38] Most cases resolve within a year [39, 40] and only a small proportion (< 15%) of stones require interventions. [40] Urinary tract infections after birth represent another risk factor for slow resolution of these calcifications.
The presence of microscopic calcification in "generic" chronic kidney disease (CKD) is relatively understudied. One study documented progressive worsening of microscopic nephrocalcinosis with worsening stages of CKD (4.6, 14.3, 20.2, and 54.0% in patients with CKD stages 1-2, 3–4, and 5, respectively). Lower serum bicarbonate level and higher serum parathyroid hormone and calcium levels represent indedendent risk factors for severity. [41]
There has been a growing awareness of the diffusely increased calcifications in patients with advanced kidney failure and end-stage kidney disease. [42] In the uremic environment, the use of large, pharmacologic doses of vitamin-D analogues and calcium-based phosphorus binders appears to accelerate the process. The presence of extraskeletal calcifications seems to be more closely correlated with the calcium-phosphorus product (sometimes referred to as the double product) and total-body calcium overload than with the presence of hypercalcemia. Calcifications in such cases are not limited to the kidneys but may involve multiple organs, including the heart, vascular beds, parenchymal organs, skin, and subcutaneous tissues.
Epidemiology
The exact epidemiology and disease burden for nephrocalcinosis is not known. Prematurity-associated calcifications are known to resolve, albeit with significant inter-individual variability. [39] Younger age in general is very suspicious for underlying genetic disorders, such as Bartter syndrome, [43, 44] Dent disease or variants of Dent disease, [45] or amelogenesis imperfecta. [35, 36] Whole-exome sequencing is a novel and very powerful technology to detect a monogenic cause of nephrocalcinosis and early-onset nephrolithiasis. [46, 47] In older age groups, acquired processes such as distal renal tubular acidosis are more likely. [11, 48]
Prognosis
The prognosis depends mainly on the etiology of the nephrocalcinosis. Patients with idiopathic hypercalciuria and medullary sponge kidney have the least risk of kidney failure and the best prognosis, whereas patients with primary type 1 hyperoxaluria have the worst prognosis. [49]
The morbidity and mortality associated with nephrocalcinosis depend on the disease associated with the condition rather than on the nephrocalcinosis itself. [50] The major long-term complication in patients with medullary nephrocalcinosis is kidney failure. Early treatment of reversible causes of kidney failure, such as urinary infections, obstruction, and hypertension, is essential. Once chronic kidney disease has developed, treatment should focus on appropriate management of chronic kidney disease and its complications.
Patient Education
In educating patients about nephrocalcinosis, key points to emphasize include the following:
-
Nephrocalcinosis is usually an incidental finding
-
Hypercalcemia or hypercalciuria are frequently present
-
Nephrocalcinosis is more likely to be a consequence of an underlying abnormality than it is to be the cause
-
Diagram of nephron.
-
Nephrocalcinosis.
-
Nephrocalcinosis.
-
Nephrocalcinosis.
-
Nonenhanced coronal CT scans through kidneys, showing cortical and medullary nephrocalcinosis (left kidney). Both kidneys appear scarred. Note thinning of renal cortex at upper pole of left kidney. Patient gave long history of chronic pyelonephritis, which is unusual cause of nephrocalcinosis.
-
Axial CT scans from patient with long history of renal tubular acidosis. Images show bilateral medullary nephrocalcinosis (early arterial phase).
-
Ultrasonogram of right kidney in woman with nephrocalcinosis. Image shows hyperechoic foci in pyramids.
-
Excretory urogram obtained at 15 minutes in man with renal papillary necrosis (most likely, patient with diabetes mellitus and repeated urinary tract infections). Image shows bilateral hydronephrosis and hydroureter due to obstruction by sloughed papillae at lower end of ureter.
-
Plain kidney-ureter-bladder (KUB) radiograph in man with renal papillary necrosis (most likely, patient with diabetes mellitus and repeated urinary tract infections). Image shows bilateral renal calcification. Large, sloughed, and calcified renal papilla is present in region of left vesicoureteric junction. Note 2 pelvic phleboliths opposite ischial spine on right.





