Background
Aortitis is histopathologic diagnosis of inflammation of the aorta, [1] and it is representative of a cluster of large-vessel diseases that have various or unknown etiologies. Although inflammation can occur in response to any injury, including trauma, the most common known causes are infections, immunologic, or connective tissue disorders. Infections can trigger a noninfectious vasculitis by generating immune complexes or by cross-reactivity. The etiology is important because immunosuppressive therapy, the main treatment for vasculitis, could aggravate an active infectious process.
Inflammation of the aorta can cause aortic dilation, resulting in aortic insufficiency. Additionally, it can cause fibrous thickening of the aorta and ostial stenosis of major branches, resulting in reduced or absent pulses and/or low blood pressure in the upper extremities, possibly with central hypertension due to renal artery stenosis. Depending on what other vessels are involved, ocular disturbances, neurological deficits, claudication, and other manifestations of vascular impairment may accompany this disorder. See the image below.
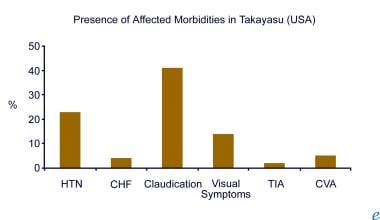 Aortitis. This chart represents the presence of an associated morbidity in Takayasu arteritis in the United States (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr).
Aortitis. This chart represents the presence of an associated morbidity in Takayasu arteritis in the United States (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr).
Agents known to infect the aorta include Neisseria (eg, gonorrhea), tuberculosis, Rickettsia (eg, Rocky Mountain spotted fever) species, spirochetes (eg, syphilis), fungi (eg, aspergillus, mucormycosis), and viruses (eg, herpes, varicella-zoster, hepatitis B, hepatitis C).
Immune disorders affecting the aorta include Takayasu arteritis, giant cell arteritis, polyarteritis nodosa, Behcet disease, Cogan syndrome, sarcoidosis, spondyloarthropathy, serum sickness, cryoglobulinemia, systemic lupus erythematosus (SLE), rheumatoid arthritis, Henoch-Schönlein purpura, and postinfectious or drug-induced immune complex disease.
Also, anti-neutrophil cytoplasmic autoantibody (ANCA) disorders can affect the large vessels, as in Wegener granulomatosis, polyangiitis, and Churg-Strauss syndrome. Other antibodies such as anti-glomerular basement membrane (ie, Goodpasture syndrome) and anti-endothelial (ie, Kawasaki disease) can also be culprits. Transplant rejection, inflammatory bowel diseases, and paraneoplastic vasculitis also may afflict the large vessels.
The European Society of Cardiology has issued guidelines on the diagnosis and treatment of aortic diseases. [2]
Pathophysiology
Aortitis has three phases. Phase I is the prepulseless inflammatory period characterized by nonspecific systemic symptoms which may include low-grade fever, fatigue, arthralgia, and weight loss, with elevated inflammation markers (sedimentation rate, C-reactive protein, plasma viscosity). Phase II involves vascular inflammation associated with pain (eg, carotidynia) and/or tenderness of the arteries. Phase III is permanent wall injury, which may present with ischemic symptoms and signs secondary to dilation, intramural tearing, narrowing, or occlusion of the proximal or distal branches of the aorta. Bruits frequently are heard, especially over carotid arteries and the abdominal aorta. The extremities may become cool, or erythematous when dependent. There may be pain with use of the extremities (ie, arm or leg claudication). Even in advanced phase III, findings may be subtle leading to postmortem diagnosis.
In advanced cases, occlusion of the vessels to the extremities may result in ischemic skin changes, ulcerations or gangrene, and with the involvement of cerebral arteries, a stroke can occur. The chronic nature of the disease promotes development of collateral circulation to the areas affected by stage III vasculitis.
The distinction between Takayasu and giant cell arteritis is primarily the clinical pattern of vessels involved. Pathologic changes involved in Takayasu arteritis are the same as for giant cell arteritis. Involved vessel walls develop irregular thickening and intimal wrinkling. Early in the disease, mononuclear infiltration with perivascular cuffing is seen. That extends to the media, followed by granulomatous changes and patches of necrosis and scarring (fibrosis) of all layers, especially the intima. Late stages have lymphocytic infiltration.
Giant cell arteritis commonly involves the temporal artery, whereas Takayasu arteritis primarily involves the aorta, its main branches, and, in 50% of cases, the pulmonary artery. [3] The initial vascular lesions with Takayasu arteritis frequently occur in or at the origin of the left subclavian artery, which can cause diminished radial pulse and easy fatigability with exertion in the left arm. As the disease progresses, the left common carotid, vertebral, brachiocephalic, right-middle or proximal subclavian, right carotid, and vertebral arteries, as well as the aorta, may be affected, as well as the retinal vessels. The image below illustrates the frequency of vascular involvement in Takayasu in patients in the United States.
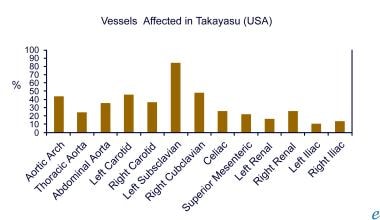 Aortitis. The frequency of vascular involvement in Takayasu arteritis is depicted (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr).
Aortitis. The frequency of vascular involvement in Takayasu arteritis is depicted (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr).
When the abdominal aorta and its branches (eg, the renal arteries) are involved, central hypertension may develop. Accurate blood pressure measurement may be difficult because of arterial lesions affecting blood flow to the extremities.
Etiology
The pathogenesis of Takayasu arteritis has not been elucidated completely. Genetic influences and immunological mechanisms have received the most attention. The associations of Takayasu arteritis with other autoimmune diseases, such as connective tissue diseases and ulcerative colitis, provide clinical support for the importance of autoimmunity in the pathogenesis.
High titers of anti-endothelial antibodies have been detected in patients with Takayasu arteritis. In a study of 19 patients by Eichorn et al, antiendothelial antibodies were found in 18, and the titers were approximately 20 times higher than normal. [4] Chauhan et al showed that the antibodies are directed against 60-65 kd antigens and may induce expression of endothelial adhesion molecules, cytokine production, and apoptosis. [5] Whether this antibody is pathogenic or merely an epiphenomenon secondary to the vascular injury remains unclear. The presence of elevated anti-cardiolipin antibody titer also has been reported.
Cell-mediated immunological mechanisms are thought to be of primary importance. Histopathologic examination has shown heavily infiltrating cells in all layers of the aorta, including alpha-beta T cells, gamma-delta T cells, and natural killer (NK) cells. In comparison to the cells found in a patient with an atherosclerotic aortic aneurysm, the proportion of gamma-delta T cells (ie, cytotoxic cells) was much higher.
Enhanced expression of human leukocyte antigen (HLA) molecules and restricted usage of alpha-beta T-cell receptor genes and gamma-delta T-cell receptor genes in the infiltrating cells suggest the existence of a targeted specific antigen. Gamma-delta T cells can recognize the major histocompatibility complex (MHC) class I (MIC) chain-related molecules MICA and MICB, whose expression is known to be increased by stress. The MICA gene was found to be located near the HLA-B gene. MICA-1.2 is strongly associated with Takayasu arteritis, even in the absence of HLA-B52, which is highly prevalent in Japanese patients. Expression of heat shock protein-65, a stress-induced protein, also is increased in the tissue. These findings suggest that unknown stress, such as infection, may trigger the autoimmune process involved in patients with Takayasu arteritis.
Epidemiology
United States data
In the United States and Europe, incidence is 1-3 new cases per year per million population. In a cohort of 1204 surgical aortic specimens described by Rojo-Leyva et al, 168 (14%) had inflammation and 52 (4.3%) were classified as having idiopathic aortitis. [6] Among 383 individuals with thoracic aortic aneurysms, 12% had idiopathic aortitis.
International data
Vasculitis has a worldwide distribution, with the greatest prevalence among Asians. An extensive epidemiological study conducted in Japan in 1984 identified 20 cases per million population. In 1990, Takayasu arteritis was added to the list of intractable diseases maintained by the Japanese Ministry of Health and Welfare; by the year 2000, 5000 patients were registered (the reported prevalence increased 2.5-fold).
Sex- and age-related demographics
Vasculitis is most common among women of reproductive age (female cases outnumber male at a ratio of 9:1).
Aortitis is most commonly discovered at age 10-40 years.
Prognosis
Morbidity/mortality
Overall, 10-year survival rate has been reported as 80%-90%.The major predictors of poor outcome are late diagnosis, complications (eg, Takayasu retinopathy, hypertension, aortic regurgitation, aneurysm) and relentless progression, which may be associated with an infectious etiology. A case report identified rapid progression of fungal aortitis in just 10 days. [7] A similar rapid progression was observed with bacterial aortitis. [8]
Patients with no complications or with mild to moderately severe complications have a 10-year survival rate of 100% and a 15-year survival rate of 93%-96%. Complications or progression reduce the 15-year survival rate to 66%-68%.
The occurrence of both a major complication and progressive course predicts the worst outcome (43% survival rate at 15-years).
Cases that are diagnosed late may enter a fulminant course leading quickly to death unless very aggressive immunotherapy is instigated promptly.
Complications
Complications include aortic insufficiency, angina pectoris, myocardial infarction, stroke, limb ischemia, renal artery hypertension, and all consequences of vascular disease.
-
Aortitis. This chart represents the presence of an associated morbidity in Takayasu arteritis in the United States (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr).
-
Aortitis. The frequency of vascular involvement in Takayasu arteritis is depicted (adapted from combined reports by Maksimowicz-McKinnon et al and Kerr).
-
Aortitis. Diffuse stenosis from the aortic arch to the abdominal aorta. The left common carotid artery is also stenotic (top arrow) and the left subclavian artery is not visualized (second arrow from the top).
-
Aortitis. Bilateral dilatation of the vertebral arteries. Occlusion of the right internal carotid artery (left arrow). Severe stenosis of the left internal carotid artery are shown. Note the moderate stenosis of the left external carotid artery at the bifurcation with dilated collateral circulation.
-
Aortitis. This image reveals moderate stenosis of the external iliac artery at the bifurcation and occlusion of the right femoral artery.
-
Aortitis. This image demonstrates leukocyte infiltration of the vasa vasorum of the aorta accompanies arteritis obliterans and ischemic necrosis of the media in a case of syphilitic aortitis.
-
Aortitis. Granulomatous arteritis with thrombosis of a cerebral vessel may present as a neurologic defect with no obvious vascular disease by history or physical examination.



