Practice Essentials
Myeloproliferative diseases are a heterogeneous group of disorders characterized by cellular proliferation of one or more hematologic cell lines in the peripheral blood, distinct from acute leukemia. The peripheral smear below shows leukoerythroblastosis and giant platelets in a patient with myelofibrosis.
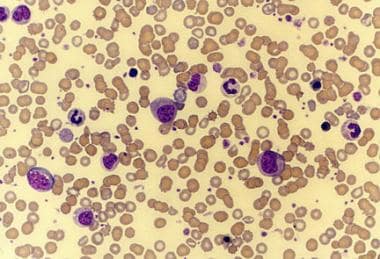 Peripheral smear of a patient with agnogenic myeloid metaplasia (myelofibrosis) shows leukoerythroblastosis. This photomicrograph also shows giant platelets.
Peripheral smear of a patient with agnogenic myeloid metaplasia (myelofibrosis) shows leukoerythroblastosis. This photomicrograph also shows giant platelets.
Patients are at risk for thrombotic and hemorrhagic events. They are also at risk of developing secondary acute leukemia from their underlying disorder, as well as from their treatment.
Signs and symptoms
Patients may have a history of the following:
-
Easy fatigability
-
Anorexia, weight loss
-
Abdominal discomfort and early satiety secondary to splenomegaly: Most common in chronic myelogenous leukemia and agnogenic myeloid metaplasia
-
Easy bruising, bleeding, and/or symptoms of thrombosis
-
Swollen, painful joint(s) secondary to gouty arthritis that is secondary to hyperuricemia
-
Priapism, tinnitus, or stupor from leukostasis
-
Left upper quadrant and left shoulder pain as a consequence of splenic infarction and perisplenitis
Clinical symptoms can include the following:
-
Pallor (except in patients with polycythemia vera)
-
Plethora secondary to polycythemia
-
Petechiae and/or ecchymosis
-
Palpable spleen and/or liver
-
Occasionally, syndrome of fever accompanied by painful, maculopapular, violaceous lesions on the trunk, arms, legs, and face; this is called acute febrile neutrophilic dermatosis, or Sweet syndrome
See Presentation for more detail.
Diagnosis
Laboratory studies
The following laboratory studies can be used in the diagnosis of myeloproliferative disease:
-
Complete blood count (CBC) and differential count with microscopic examination of the peripheral smear
-
Leukocyte alkaline phosphatase (LAP) score: To differentiate chronic myelogenous leukemia from other causes of leukocytosis
-
Polymerase chain reaction (PCR) assay or fluorescent in-situ hybridization (FISH) run on peripheral blood: Can detect bcr-abl gene rearrangement; this helps to differentiate chronic myelogenous leukemia from other myeloproliferative diseases
-
Red blood cell mass study: True versus spurious polycythemia
-
Serum uric acid level
-
PCR assay run on bone marrow: To test for JAK2; available for suspected cases of polycythemia vera, essential thrombocythemia, and myelofibrosis
Biopsy
Bone marrow aspiration and biopsy with cytogenetic studies are required in most, but not all, patients. Cytogenetic studies detect the presence or absence of the Philadelphia chromosome and help to differentiate myeloproliferative disorders from myelodysplastic syndrome.
Bone marrow histology shows hypercellularity in most of these disorders. In the case of myelofibrosis, bone marrow fibrosis is demonstrated on the reticulin stain. Bone marrow fibrosis is also detected in the spent phase of chronic myelogenous leukemia and polycythemia vera.
See Workup for more detail.
Management
Chronic myelogenous leukemia
Hematopoietic stem cell transplantation can be considered in young patients with chronic myelogenous leukemia in chronic phase if a human leukocyte antigen (HLA)-matched donor is available.
Agents used in the treatment of the disease include the following:
-
Imatinib mesylate (Gleevec): Approved for use in Philadelphia chromosome–positive chronic myelogenous leukemia patients in chronic phase; also indicated for chronic myelogenous leukemia in blast crisis, accelerated phase, or in chronic phase after interferon-alfa therapy failure; this is the treatment of choice for most patients. [1]
-
Interferon alfa: Produces hematologic and molecular remissions in some patients with chronic myelogenous leukemia
-
Low-dose cytosine arabinoside: When added to interferon alfa, has been reported to increase remission rates.
-
Hydroxyurea: For patients with chronic myelogenous leukemia who are intolerant to interferon-alfa therapy
-
Dasatinib (Sprycel): Indicated for the treatment of adult patients with chronic myeloid leukemia in chronic, accelerated, or myeloid or lymphoid blast phase who are resistant or intolerant to prior therapy including imatinib.
-
Nilotinib (Tasigna): Indicated for the treatment of chronic-phase and accelerated-phase Philadelphia chromosome-positive chronic myelogenous leukemia in adult patients who are resistant or intolerant to prior therapy including imatinib
Polycythemia vera
Treatment for this disease is palliative. Young (< 40 y), asymptomatic patients with polycythemia vera can be considered for therapeutic phlebotomies alone to maintain a hematocrit level of less than 45%. Other patients can undergo myelosuppressive therapy with hydroxyurea. Radioactive phosphorous can be used as an alternative therapy in older patients.
Essential thrombocythemia
No curative treatment is available. The aim of therapy is to maintain the patient’s platelet count within the reference range. This usually can be achieved with hydroxyurea or anagrelide.
Myelofibrosis
Asymptomatic patients can be monitored clinically until symptomatic. Hydroxyurea is useful to suppress the number of circulating cells.
Patients with painful, massively enlarged spleens refractory to myelosuppressive therapy are occasionally treated with radiation therapy, but they may ultimately require splenectomy.
In November 2011, a JAK1/JAK2 inhibitor, ruxolitinib (Jakafi), became the first US Food and Drug Administration (FDA)–approved drug for patients with intermediate- or high-risk myelofibrosis.
Allogeneic hematopoietic stem cell transplantation (ASCT) is curative for primary myelofibrosis (PMF) and may be an alternative for selected patients whose disease is refractory to conventional therapies.
See Treatment and Medication for more detail.
Background
Myeloproliferative diseases (MPDs) are a heterogeneous group of disorders characterized by cellular proliferation of one or more hematologic cell lines in the peripheral blood, distinct from acute leukemia.
According to the French-American-British (FAB) classification, chronic myeloproliferative diseases consist of the following four diseases:
-
Chronic myelogenous leukemia (CML), shown in the image below
-
Polycythemia vera (PV)
-
Primary myelofibrosis (MF), previously known as agnogenic myeloid metaplasia (AMM)
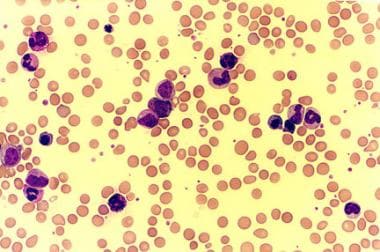 Peripheral smear of a patient with chronic myelogenous leukemia (CML) shows leukocytosis with extreme left shift and basophilia.
Peripheral smear of a patient with chronic myelogenous leukemia (CML) shows leukocytosis with extreme left shift and basophilia.
In 2002, the World Health Organization (WHO) proposed an alternate classification schema for these diseases, adding chronic neutrophilic leukemia (CNL) and chronic eosinophilic leukemia (CEL)/hypereosinophilic syndrome (HES). [2] The WHO updated its classification in 2008 and changed the nomenclature from “chronic myeloproliferative disease” to “myeloproliferative neoplasms”. [3] In 2016, the WHO further revised its classification of hematopoietic tumors and now recognizes several major categories of myeloid malignancies including myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN), MDS/MPN overlap, mastocytosis, eosinophilia‐associated myeloid/lymphoid neoplasms with specific mutations (e.g., PDGFR) and myeloid neoplasms with germline predisposition. [4] For a comparison of the FAB and 2016 WHO classification systems, see the table below.
Table. Comparison of FAB and WHO Classifications of Myeloproliferative Neoplasms (MPN) (Open Table in a new window)
FAB |
WHO |
Chronic myelogenous leukemia |
Chronic myelogenous leukemia, BCR/ABL1 positive |
Polycythemia vera |
Polycythemia vera |
Essential thrombocythemia |
Essential thrombocythemia |
Agnogenic myeloid metaplasia/myelofibrosis |
Primary myelofibrosis* |
... |
Chronic neutrophilic leukemia, not otherwise specified |
... |
MPN, unclassified |
| *The 2016 WHO classification system distinguishes prefibrotic (prePMF) from overtly fibrotic PMF [4] | |
In some patients, conditions overlap, and clear categorization may be difficult. Myeloproliferative disease may evolve into one of the other myeloproliferative conditions, transform to acute leukemia, or both.
Some evidence indicates that myeloproliferative diseases arise from malignant transformation of a single stem cell. Involvement of erythropoiesis, neutrophilopoiesis, eosinophilopoiesis, basophilopoiesis, monocytopoiesis, and thrombopoiesis occurs in the chronic phase of CML. Some evidence also indicates that lymphocytes are derived from primordial malignant cells. This is based on observations that a single isoenzyme for glucose-6-phosphate dehydrogenase (G6PD) is present in some T and B lymphocytes in women with CML who are heterozygous for isoenzymes A and B.
Pathophysiology
Data from glucose-6-phosphate dehydrogenase (G6PD) studies, cytogenetic analyses, and molecular methods have established the clonal origin of myeloproliferative diseases; this clonality potentially occurs at different stem cell levels. An attribute common to these disorders appears to be an acquired activating mutation in the gene coding for various tyrosine kinases.
In chronic myelogenous leukemia, the tyrosine kinase activity of the bcr-abl hybrid gene is increased. In polycythemia vera, essential thrombocythemia, and myelofibrosis (see the following images), the prevalent genetic lesion appears to be a valine to phenylalanine substitution at amino acid position 617 (V617F) within the Janus kinase 2 (JAK2) gene. [5, 6, 7, 8] This produces hypersensitivity to erythropoietin. At least in myelofibrosis patients the leukemic transformation is probably not related to JAK-2 (V617F) mutation status. [9]
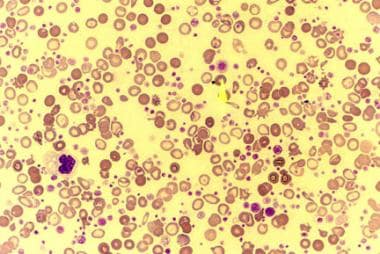 Peripheral smear of a patient with essential thrombocythemia (ET) shows markedly increased number of platelets. Some of the platelets are giant (arrow).
Peripheral smear of a patient with agnogenic myeloid metaplasia (myelofibrosis) shows leukoerythroblastosis. This photomicrograph also shows giant platelets.
Peripheral smear of a patient with essential thrombocythemia (ET) shows markedly increased number of platelets. Some of the platelets are giant (arrow).
Peripheral smear of a patient with agnogenic myeloid metaplasia (myelofibrosis) shows leukoerythroblastosis. This photomicrograph also shows giant platelets.
A study by Anand et al found that JAK2 mutations generate expansion of later myeloid differentiation compartments, in which homozygous expression of the mutation confers an added proliferative advantage at the single-cell level. The findings suggest that JAK2 inhibitors may control myeloproliferation; however, they may have limited efficacy in eradicating leukemic stem cells. [10]
Systemic mastocytosis has been linked with the D816 mutation of the KIT gene. The FIP1L1-PDGFR mutation has been identified in a subgroup of people with systemic mastocytosis with eosinophilia (SM-eos). [11]
Nangalia et al reported that somatic mutations in the CALR gene exist in the majority of patients without JAK2 and MPL mutations, who comprise about 68% of patients with essential thrombocythemia and 88% of patients with primary myelofibrosis. On clonal analyses, CALR mutations were found in the earliest phylogenetic node, a finding consistent with its role as a possible initiating mutation. [12, 13, 14]
Etiology
As with other malignant disorders, the precise cause of myeloproliferative disease is unknown. The etiology is complex, incompletely understood, and likely a multistep process involving more than one gene.
Philadelphia chromosome, t(9:22), is found in most patients who have chronic myelogenous leukemia. Even when Philadelphia chromosome testing is negative, the gene bcr-abl, formed as result of t(9:22), tests positive in patients with chronic myelogenous leukemia using molecular techniques. Bcr-abl encodes a fusion protein with tyrosine kinase activity, which is constitutively expressed and is regarded as the central mechanism that underlies the chronic phase of chronic myelogenous leukemia. [15]
In a retrospective study of 11,000 patients in Sweden with myeloproliferative neoplasm, the authors reviewed the incidence of acute myeloid leukemia (AML) and myelodysplastic syndrome that were secondary to treatment; the study found that treatment with radioactive phosphorus but not hydroxyurea increased the risk of myelodysplastic syndrome and AML. [16]
Epidemiology
The American Cancer Society estimates that 8990 new cases of chronic myelogenous leukemia (CML) will be diagnosed in the United States in 2019. [17] CML accounts for more than half of myeloproliferative disease cases. The incidence of polycythemia vera in the United States is approximately 5-17 cases per 1 million population per year. True incidences of essential thrombocythemia and myelofibrosis are not known because epidemiological studies on these disorders are inadequate.
The incidence of polycythemia vera is 0.02-2.8 per 100,000 per year [18, 19] ; Japan has the lowest incidence. Essential thrombocythemia has an incidence of 0.1-1.5 per 100,000 per year. Myelofibrosis has an international incidence of 0.4-0.9 per 100,000 per year.
CML appears to affect all races with approximately equal frequency. In a study from northern Israel, the incidences of polycythemia vera, essential thrombocythemia, and myelofibrosis in Ashkenazi Jews were 10-fold higher than in Sephardic Jews, and 20-fold higher than in Arabs. [3]
The female-to-male ratio is 1:1.4. The American Cancer Society estimates that new cases of CML will be diagnosed in 5250 males and 3740 females in 2019. [17]
Most cases encountered in clinical practice are in patients aged 40-60 years. Myeloproliferative diseases are uncommon in people younger than 20 years and are rare in childhood.
Prognosis
The current (2008-2014) 5-year relative survival rate for adults (ages 20 and older) is 67% for CML. Survival rates for CML hav more than tripled, up from 22% in the mid-1970s, in large part due to the discovery and use of targeted drugs over the past two decades. The American Cancer Society estimates that 1040 deaths from chronic myelogenous leukemia (CML) will occur in 2019. [17]
Exact mortality and morbidity rates of other myeloproliferative diseases are unknown.
Patients are at risk of both thrombotic and hemorrhagic events. Thrombotic and hemorrhagic events are particularly common in polycythemia vera (PV) and essential thrombocythemia (ET). Thrombosis is the cause of death in 30-40% of patients. A particularly serious thrombotic event that may be associated with polycythemia vera is Budd-Chiari syndrome, which is due to hepatic venous or inferior venal caval thrombosis.
Patients are at risk of developing secondary acute leukemia from their underlying disorder as well as their treatment.
-
Peripheral smear of a patient with chronic myelogenous leukemia (CML) shows leukocytosis with extreme left shift and basophilia.
-
Peripheral smear of a patient with chronic myelogenous leukemia (CML) in blastic phase shows several blasts.
-
Peripheral smear of a patient with essential thrombocythemia (ET) shows markedly increased number of platelets. Some of the platelets are giant (arrow).
-
Peripheral smear of a patient with agnogenic myeloid metaplasia (myelofibrosis) shows leukoerythroblastosis. This photomicrograph also shows giant platelets.
-
Photomicrograph of a peripheral smear of a patient with agnogenic myeloid metaplasia (myelofibrosis) shows findings of leukoerythroblastosis, giant platelets, and few teardrop cells.
Tables
FAB |
WHO |
Chronic myelogenous leukemia |
Chronic myelogenous leukemia, BCR/ABL1 positive |
Polycythemia vera |
Polycythemia vera |
Essential thrombocythemia |
Essential thrombocythemia |
Agnogenic myeloid metaplasia/myelofibrosis |
Primary myelofibrosis* |
... |
Chronic neutrophilic leukemia, not otherwise specified |
... |
MPN, unclassified |
| *The 2016 WHO classification system distinguishes prefibrotic (prePMF) from overtly fibrotic PMF [4] | |








