Practice Essentials
Metabolic alkalosis is defined as elevation of the body's pH above 7.45. [1] Metabolic alkalosis involves a primary increase in serum bicarbonate (HCO3-) concentration, due to a loss of H+ from the body or a gain in HCO3-. As a compensatory mechanism, metabolic alkalosis leads to alveolar hypoventilation with a rise in arterial carbon dioxide tension (PaCO2), which diminishes the change in pH that would otherwise occur.
Normally, arterial PaCO2 increases by 0.5-0.7 mm Hg for every 1 mEq/L increase in plasma bicarbonate concentration, a compensatory response that is very quick. If the change in PaCO2 is not within this range, then a mixed acid-base disturbance occurs. For example, if the increase in PaCO2 is more than 0.7 times the increase in bicarbonate, then metabolic alkalosis coexists with primary respiratory acidosis. Likewise, if the increase in PaCO2 is less than the expected change, then a primary respiratory alkalosis is also present.
The first clue to metabolic alkalosis is often an elevated bicarbonate concentration that is observed when serum electrolyte measurements are obtained. Remember that an elevated serum bicarbonate concentration may also be observed as a compensatory response to primary respiratory acidosis. However, a bicarbonate concentration greater than 35 mEq/L is almost always caused by metabolic alkalosis.
Metabolic alkalosis is diagnosed by measuring serum electrolytes and arterial blood gases. If the etiology of metabolic alkalosis is not clear from the clinical history and physical examination, including drug use and the presence of hypertension, then a urine chloride ion concentration can be obtained. Calculation of the serum anion gap may also help to differentiate between primary metabolic alkalosis and metabolic compensation for respiratory acidosis. (See Workup.)
The management of metabolic alkalosis depends primarily on the underlying etiology and on the patient’s volume status. Direct treatment of the alkalosis itself (eg, administration of acidic intravenous solutions) may be indicated in some cases (see Treatment).
An algorithmic approach to metabolic alkalosis is depicted in the image below.
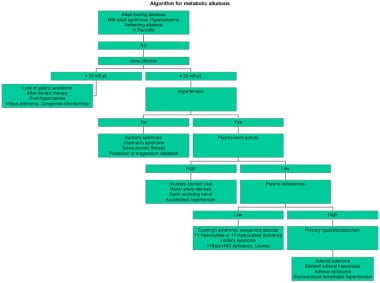 Algorithm for metabolic alkalosis.
Algorithm for metabolic alkalosis.
For a discussion of metabolic alkalosis in children, see Pediatric Metabolic Alkalosis. For a general review of acid-base regulation, see Metabolic Acidosis.
Pathophysiology
The organ systems involved in metabolic alkalosis are mainly the kidneys and GI tract. The pathogenesis involves two processes, the generation of metabolic alkalosis and the maintenance of metabolic alkalosis, which usually overlap.
Generation of metabolic alkalosis
Metabolic alkalosis may be generated by one of the following mechanisms:
-
Loss of hydrogen ions
-
Shift of hydrogen ions into the intracellular space
-
Alkali administration
-
Contraction alkalosis
Hydrogen ions may be lost through the kidneys or the GI tract. Vomiting or nasogastric (NG) suction generates metabolic alkalosis by the loss of gastric secretions, which are rich in hydrochloric acid (HCl). Whenever a hydrogen ion is excreted, a bicarbonate ion is gained in the extracellular space.
Renal losses of hydrogen ions occur whenever the distal delivery of sodium increases in the presence of excess aldosterone, which stimulates the electrogenic epithelial sodium channel (ENaC) in the collecting duct. As this channel reabsorbs sodium ions, the tubular lumen becomes more negative, leading to the secretion of hydrogen ions and potassium ions into the lumen.
Shift of hydrogen ions into the intracellular space mainly develops with hypokalemia. As the extracellular potassium concentration decreases, potassium ions move out of the cells. To maintain neutrality, hydrogen ions move into the intracellular space.
Administration of sodium bicarbonate in amounts that exceed the capacity of the kidneys to excrete this excess bicarbonate may cause metabolic alkalosis. This capacity is reduced when a reduction in filtered bicarbonate occurs, as observed in renal failure, or when enhanced tubular reabsorption of bicarbonate occurs, as observed in volume depletion (see Maintenance of metabolic alkalosis).
Loss of bicarbonate-poor, chloride-rich extracellular fluid, as observed with thiazide diuretic or loop diuretic therapy or chloride diarrhea, leads to contraction of extracellular fluid volume. Because the original bicarbonate mass is now dissolved in a smaller volume of fluid, an increase in bicarbonate concentration occurs. This increase in bicarbonate causes, at most, a 2- to 4-mEq/L rise in bicarbonate concentration.
Maintenance of metabolic alkalosis
Decreased perfusion to the kidneys, caused by either volume depletion or a reduction in effective circulating blood volume (eg, edematous states such as heart failure or cirrhosis) stimulates the renin-angiotensin-aldosterone system. This increases renal sodium ion reabsorption throughout the nephron, including the principal cells of the collecting duct, and results in enhanced hydrogen ion secretion via the apical proton pump H+ adenosine triphosphatase (ATPase) in the adjacent A-type intercalated cells.
Aldosterone may also independently increase the activity of the apical proton pump in the collecting duct. Whenever a hydrogen ion is secreted into the tubular lumen, a bicarbonate ion is gained into the systemic circulation via the basolateral Cl-/HCO3- exchanger.
Chloride depletion may occur through the GI tract by loss of gastric secretions, which are rich in chloride ions, or through the kidneys with loop diuretics or thiazides. Chloride depletion, even without volume depletion, enhances bicarbonate reabsorption by different mechanisms as discussed below.
In the late thick ascending limb (TAL) and early distal tubule, specialized cells called the macula densa are present. These cells have an Na+/K+/2Cl- cotransporter in the apical membrane, which is mainly regulated by chloride ions. When fewer chloride ions reach this transporter (eg, chloride depletion), the macula densa signals the juxtaglomerular apparatus (ie, specialized cells in the wall of the adjacent afferent arteriole) to secrete renin, which increases aldosterone secretion via angiotensin II.
In alkalemia, the kidneys secrete the excess bicarbonate via the apical chloride/bicarbonate exchanger, pendrin, in the B-type intercalated cells of the collecting duct. In this way, protons are gained to the systemic circulation via the basolateral H+ ATPase. In chloride depletion, fewer chloride ions are available to be exchanged with bicarbonate, and the ability of the kidneys to excrete the excess bicarbonate is impaired.
Many of the causes of metabolic alkalosis are also associated with hypokalemia. In turn, hypokalemia maintains metabolic alkalosis by five different mechanisms.
First, hypokalemia results in the shift of hydrogen ions intracellularly. The resulting intracellular acidosis enhances bicarbonate reabsorption in the collecting duct.
Second, hypokalemia stimulates the apical H+/K+ ATPase in the collecting duct. Increased activity of this ATPase leads to teleologically appropriate potassium ion reabsorption but a corresponding hydrogen ion secretion. This leads to a net gain of bicarbonate, maintaining systemic alkalosis.
Third, hypokalemia stimulates renal ammonia genesis, reabsorption, and secretion. Ammonium ions (NH4+) are produced in the proximal tubule from the metabolism of glutamine. During this process, alpha-ketoglutarate is produced, the metabolism of which generates bicarbonate that is returned to the systemic circulation. Hypokalemia stimulates NH4+ uptake via the Na+/K+/2Cl- cotransporter of TAL because NH4+ competes with K+ for the transporter. Hypokalemia increases the expression of the ammonia transporter RhBG, which increases NH3 excretion in the collecting duct.
Fourth, it leads to impaired chloride ion reabsorption in the distal nephron. This results in an increase in luminal electronegativity, with subsequent enhancement of hydrogen ion secretion.
Fifth, it reduces the glomerular filtration rate (GFR). Animal studies have shown that hypokalemia, by unknown mechanisms, decreases GFR, which decreases the filtered load of bicarbonate. In the presence of volume depletion, this impairs renal excretion of the excess bicarbonate.
Etiology
The most common causes of metabolic alkalosis are the use of diuretics and the external loss of gastric secretions. Causes of metabolic alkalosis can be divided into chloride-responsive alkalosis (urine chloride < 20 mEq/L), chloride-resistant alkalosis (urine chloride > 20 mEq/L), and other causes, including alkali-loading alkalosis.
Chloride-responsive alkalosis
The principal causes of chloride-responsive alkalosis are the loss of gastric secretions, ingestion of large doses of nonabsorbable antacids, and use of thiazide or loop diuretics. [2] Miscellaneous causes account for the remainder of cases. [3]
Gastric secretions are rich in hydrochloric acid (HCl). The secretion of HCl by the stomach usually stimulates bicarbonate secretion by the pancreas once the HCl reaches the duodenum. Ordinarily, these pancreatic secretions neutralize the gastric secretions, and no net gain or loss of hydrogen ions or bicarbonate occurs.
When HCl is lost through vomiting (including purging, in persons with eating disorders [4] ) or nasogastric suction, pancreatic secretions are not stimulated and a net gain of bicarbonate into the systemic circulation occurs, generating a metabolic alkalosis. Volume depletion maintains alkalosis. In this case, the hypokalemia is secondary to the alkalosis itself and to renal loss of potassium ions from the stimulation of aldosterone secretion.
Ingestion of large doses of nonabsorbable antacids (eg, magnesium hydroxide) may generate metabolic alkalosis by a rather complicated mechanism. Upon ingestion of magnesium hydroxide, calcium, or aluminum with base hydroxide or carbonate, the hydroxide anion buffers hydrogen ions in the stomach. The cation binds to bicarbonate secreted by the pancreas, leading to loss of bicarbonate with stools. In this process, both hydrogen ions and bicarbonate are lost, and, usually, no acid-base disturbance occurs. Sometimes, however, not all the bicarbonate binds to the ingested cation, which means that some bicarbonate is reabsorbed in excess of the lost hydrogen ions. This occurs primarily when antacids are administered with a cation-exchange resin (eg, sodium polystyrene sulfonate [Kayexalate]); the resin binds the cation, leaving bicarbonate unbound.
Thiazides and loop diuretics enhance sodium chloride excretion in the distal convoluted tubule and the thick ascending loop, respectively. These agents cause metabolic alkalosis by chloride depletion and by increased delivery of sodium ions to the collecting duct, which enhances potassium ion and hydrogen ion secretion.
Volume depletion also stimulates aldosterone secretion, which enhances sodium ion reabsorption in the collecting duct and increases hydrogen ion and potassium secretion in this segment. Urine chloride is low after discontinuation of diuretic therapy, while it is high during active diuretic use.
Miscellaneous causes of metabolic alkalosis include villous adenomas, which are a rare cause of diarrhea. Villous adenomas usually lead to metabolic acidosis from loss of colonic secretions that are rich in bicarbonate, but occasionally these tumors cause metabolic alkalosis. The mechanism is not well understood. Some authors opine that hypokalemia from these tumors is the etiology of the metabolic alkalosis.
Congenital chloridorrhea (see Chloride Diarrhea, Familial. Online Mendelian Inheritance in Man [OMIM]) is a rare form of severe secretory diarrhea that is inherited as an autosomal recessive trait. Mutations in the down-regulated adenoma gene result in defective function of the chloride/bicarbonate exchange in the colon and ileum, leading to increased secretion of chloride and reabsorption of bicarbonate.
During respiratory acidosis, the kidneys reabsorb bicarbonate and secrete chloride to compensate for the acidosis. In the posthypercapnic state, urine chloride is high and can lead to chloride depletion. Once the respiratory acidosis is corrected, the kidneys cannot excrete the excess bicarbonate because of the low luminal chloride.
Infants with cystic fibrosis may develop metabolic alkalosis because of loss of chloride in sweat. These infants are also prone to volume depletion.
Chloride-resistant alkalosis
Causes of chloride-resistant alkalosis can be divided into those associated with hypertension and those associated with hypotension or normotension. The former may result from primary hyperaldosteronism, as well as a variety of acquired and hereditary disorders. An adrenal adenoma (most common), bilateral adrenal hyperplasia, or an adrenal carcinoma may cause primary hyperaldosteronism. [5]
Another cause of primary hyperaldosteronism is glucocorticoid-remediable aldosteronism (see Hyperaldosteronism, Familial, Type 1 [OMIM]), an autosomal dominant disorder, in which ectopic production of aldosterone in the zona fasciculata of the adrenal cortex occurs. In this case, aldosterone production is controlled by adrenocorticotropic hormone (ACTH) rather than angiotensin II and potassium, its principal regulators. This type of primary hyperaldosteronism responds to glucocorticoid therapy, which inhibits aldosterone secretion by suppressing ACTH.
The mineralocorticoid receptor (MR) in the collecting duct usually is responsive to both aldosterone and cortisol. Cortisol has a higher affinity for the MR and circulates at a higher concentration than aldosterone. Under physiological conditions, however, the enzyme 11-beta-hydroxysteroid dehydrogenase type 2 (11B-HSD2) inactivates cortisol to cortisone in the collecting duct, allowing aldosterone free access to its receptor.
Deficiency of this enzyme leads to occupation and activation of the MR by cortisol, which, like aldosterone, then stimulates the ENaC. Cortisol behaves as a mineralocorticoid under these circumstances.
11B-HSD2 deficiency may be inherited as an autosomal recessive trait (see Cortisol 11-Beta-Ketoreductase Deficiency [OMIM]), manifesting as syndrome of apparent mineralocorticoid excess (AME). The enzyme may be inhibited by glycyrrhizic acid, which is found in licorice and chewing tobacco, or carbenoxolone, which is a synthetic derivative of glycyrrhizinic acid. Deficiency or inhibition of 11B-HSD2 causes hypertension with low renin and low aldosterone, hypokalemia, and metabolic alkalosis. Serum cortisol is within the reference range because the negative feedback of cortisol on adrenocorticotropic hormone (ACTH) is intact.
Active use of thiazides or loop diuretics in hypertension is the most common cause of metabolic alkalosis in hypertensive patients. The mechanism of alkalosis is discussed above.
The enhanced mineralocorticoid effect in Cushing syndrome is caused by occupation of the MR by the high concentration of cortisol. Hypokalemia and metabolic alkalosis are more common in Cushing syndrome caused by ectopic ACTH production (90%) than in other causes of Cushing syndrome (10%). This difference is related to the higher concentration of plasma cortisol and the defective 11B-HSD activity found in ectopic ACTH production.
Liddle syndrome (see Liddle Syndrome [OMIM]) is a rare autosomal dominant disorder arising from a gain-of-function mutation in the beta (SCNN1B) or gamma subunit (SCNN1G) of the ENaC in the collecting duct. The channel behaves as if it is permanently open, and unregulated reabsorption of Na+ occurs, leading to volume expansion and hypertension. This unregulated Na+ reabsorption is responsible for secondary renal hydrogen ion and potassium ion losses and persists despite suppression of aldosterone.
Significant unilateral or bilateral renal artery stenosis stimulates the renin-angiotensin-aldosterone system, leading to hypertension and hypokalemic metabolic alkalosis.
Renin- or deoxycorticosterone-secreting tumors are rare. In renin-secreting tumors, excessive amounts of renin are secreted by tumors in the juxtaglomerular apparatus, stimulating aldosterone secretion. In the latter, deoxycorticosterone (DOC), rather than aldosterone, is secreted by some adrenal tumors and has mineralocorticoid effects.
Mutation in mineralocorticoid receptor (see Hypertension, Early-Onset, Autosomal Dominant, with Severe Exacerbation in Pregnancy [OMIM]) is a form of early-onset hypertension with autosomal dominant inheritance that has now been linked to a specific mutation of the MR. This mutation results in constitutive activation of the MR, making the MR responsive to progesterone.
Activation of MR leads to unregulated sodium ion reabsorption via the collecting duct sodium ion channel, with accompanying hypokalemia and alkalosis. The disease is characterized by severe exacerbations of hypertension during pregnancy, and spironolactone can exacerbate hypertension.
Congenital adrenal hyperplasia (CAH; see Adrenal Hyperplasia, Congenital, Due to 11-Beta-Hydroxylase Deficiency [OMIM] and Adrenal Hyperplasia, Congenital, Due to 17-Alpha-Hydroxylase Deficiency [OMIM]) can be caused by deficiency of either 11-beta-hydroxylase or 17-alpha-hydroxylase. Both enzymes are involved in the synthesis of adrenal steroids.
Deficiency of either enzyme leads to increased levels of the mineralocorticoid 11-deoxycortisol, while cortisol and aldosterone production is impaired. 11-Hydroxylase deficiency differs from 17-hydroxylase deficiency by the presence of virilization.
Chloride-resistant alkalosis (urine chloride >20 mEq/L) with hypotension or normotension may be a manifestation of Bartter syndrome (see Hypokalemic Alkalosis with Hypercalciuria [OMIM]), an inherited autosomal recessive disorder. In Bartter syndrome, impaired reabsorption of sodium ions and chloride ions in the thick ascending loop of Henle leads to their increased delivery to the distal nephron.
The impaired reabsorption of sodium chloride in the loop of Henle is secondary to loss of function mutations of 1 of several transporters in this site of the nephron: (1) the furosemide-sensitive Na+/K+/2Cl- cotransporter (NKCC2); (2) the basolateral chloride ion channel (CLCNKB); (3) the inwardly rectifying apical potassium ion channel (ROMK1); (4) barttin (BSND), the beta-subunit of the chloride channels, CLC-Ka and CLC-Kb; and (5) the calcium sensing receptor (CaSR).
Mutations of CLCNKB cause classic Bartter syndrome, while mutations of the other 2 transporters manifest with the antenatal form of Bartter syndrome. [6] Edema and hypertension are absent, and hypercalciuria is common because the impaired reabsorption of sodium chloride inhibits the paracellular reabsorption of calcium. Because loop diuretics inhibit the Na+/K+/2Cl- transporter, the electrolyte abnormalities observed in Bartter syndrome and with loop diuretic use are similar.
Gitelman syndrome (see Potassium and Magnesium Depletion [OMIM]) is an inherited autosomal recessive disorder in which loss of function of the thiazide-sensitive sodium/chloride transporter (NCCT) in the distal convoluted tubule occurs. The subsequent increased distal solute delivery and salt wasting with stimulation of the renin-angiotensin-aldosterone system lead to hypokalemic metabolic alkalosis. Other features of the syndrome are hypocalciuria and hypomagnesemia. The electrolyte abnormalities resemble those caused by thiazide diuretics.
Pure hypokalemia (ie, severe potassium ion depletion) causes mild metabolic alkalosis, but, in combination with hyperaldosteronism, the alkalosis is more severe. Possible mechanisms of alkalosis in hypokalemia are enhanced proximal bicarbonate reabsorption, stimulated renal ammonia genesis, impaired renal chloride reabsorption, reduced GFR (in animals), and intracellular acidosis in the distal nephron with subsequent enhanced hydrogen secretion.
Magnesium depletion (ie, hypomagnesemia) may lead to metabolic alkalosis. The mechanism probably involves hypokalemia, which is usually caused by or associated with magnesium depletion.
Other causes
The kidneys are able to excrete any excess alkali load, whether it is exogenous (eg, infusion of sodium bicarbonate) or endogenous (eg, metabolism of lactate to bicarbonate in lactic acidosis). However, in renal failure or in any condition that maintains the alkalosis, this natural ability of the kidneys to excrete the excess bicarbonate is impaired. Examples include the following:
-
Alkali-loading alkalosis
-
Hypercalcemia
-
Intravenous penicillin
-
Hypoproteinemic alkalosis
Milk-alkali syndrome comprises hypercalcemia, renal insufficiency, and metabolic alkalosis. Before the advent of H2-receptor antagonists, milk-alkali syndrome was observed in patients who ingested large amounts of milk and antacids as treatment for peptic ulcers. Currently, the syndrome is observed mainly in people who chronically ingest large doses of calcium carbonate, with or without vitamin D (typically for osteoporosis prevention). [7]
The hypercalcemia that develops in some of these persons increases renal bicarbonate reabsorption. Renal insufficiency can occur secondary to nephrocalcinosis or hypercalcemia and contributes to maintaining the metabolic alkalosis.
Patients with end-stage renal disease (ESRD) are dialyzed with a high concentration of bicarbonate in the dialysate to reverse metabolic acidosis (ie, hemodialysis using high bicarbonate dialysate). Sometimes, this high bicarbonate exceeds the amount needed to buffer the acidosis. Because the ability of the kidneys to excrete the excess bicarbonate is absent or severely diminished, the alkalosis persists temporarily. The degree of alkalosis might be severe if the patient also has vomiting.
Metabolic alkalosis has been reported after regional citrate anticoagulation in hemodialysis or in continuous renal replacement therapies. Citrate is infused in the blood inflow line in the hemodialysis circuit, where it prevents clotting by binding calcium. Because the dialyzer does not remove citrate completely, a fraction of the infused citrate might reach the systemic circulation. Citrate in the blood is metabolized to bicarbonate in the liver. The accumulated bicarbonate may lead to metabolic alkalosis.
In an international prospective cohort study involving 17,031 patients receiving thrice-weekly hemodialysis, high dialysate bicarbonate, especially in patients with prolonged exposure, contributed to higher mortality, most likely through development of post-dialysis metabolic alkalosis. The positive association between dialysate bicarbonate concentration and mortality (adjusted hazard ratio, 1.08 per 4 mEq/L higher; 95% confidence index [CI], 1.01–1.15; adjusted hazard ratio for dialysate bicarbonate ≥38 vs. 33–37 mEq/L, 1.07 [95% CI, 0.97–1.19]) was consistent across pre-dialysis session serum bicarbonate levels and between facilities that used one dialysate bicarbonate concentration and those that prescribed different concentrations for each patient. [8]
Metabolic alkalosis may be a potential complication of plasmapheresis in patients with renal failure. The source of alkali is the citrate that is used to prevent clotting in the extracorporeal circuit and in the stored blood from which the fresh frozen plasma is prepared. Using heparin as the anticoagulant and using albumin instead of fresh frozen plasma as the replacement solution can prevent the metabolic alkalosis.
Recovery from lactic or ketoacidosis in the presence of volume depletion or renal failure typically occurs when exogenous bicarbonate is administered to correct the acidosis. When the patient recovers, the beta-hydroxybutyrate and lactate are metabolized to bicarbonate and the original bicarbonate deficit is recovered. The administered bicarbonate now becomes a surplus.
Refeeding with a carbohydrate-rich diet after prolonged fasting results in mild metabolic alkalosis because of enhanced metabolism of ketoacids to bicarbonate.
Massive blood transfusion results in mild metabolic alkalosis as the citrate in the transfused blood is converted to bicarbonate. Metabolic alkalosis is more likely to develop in the presence of renal insufficiency.
Hypercalcemia may cause metabolic alkalosis by volume depletion and enhanced bicarbonate reabsorption in the proximal tubule. However, hypercalcemia from primary hyperparathyroidism is usually associated with a metabolic acidosis.
The intravenous administration of penicillin, carbenicillin, or other semisynthetic penicillins may cause hypokalemic metabolic alkalosis. This occurs because of distal delivery of nonreabsorbable anions with an absorbable cation such as Na+.
Metabolic alkalosis has been reported in patients with hypoproteinemia. The mechanism of alkalosis is not clear, but it may be related to loss of negative charges of albumin. A decrease in plasma albumin of 1 g/dL is associated with an increase in plasma bicarbonate of 3.4 mEq/L.
High rates of acid-base disorders have been reported in patients hospitalized with COVID-19. In one retrospective series of 211 patients, pH variations occurred in 79.7% of patients, with the most common finding being metabolic alkalosis, in a third of patients. [9] A similar rate of metabolic alkalosis was reported in an observational study of 104 hospitalized COVID-19 patients with acute respiratory distress syndrome (ARDS). [10]
Summary of causes of metabolic alkalosis
Causes of chloride-responsive alkalosis (urine chloride < 20 mEq/L) include the following:
-
Loss of gastric secretions - Vomiting, nasogastric suction
-
Loss of colonic secretions - Congenital chloridorrhea, villous adenoma
-
Thiazides and loop diuretics (after discontinuation)
-
Posthypercapnia
-
Cystic fibrosis
Causes of chloride-resistant alkalosis (urine chloride > 20 mEq/L) with hypertension include the following:
-
Primary hyperaldosteronism - Adrenal adenoma, bilateral adrenal hyperplasia, adrenal carcinoma, glucocorticoid-remediable hyperaldosteronism
-
11B-HSD2 - Genetic, licorice, chewing tobacco, carbenoxolone
-
CAH - 11-Hydroxylase or 17-hydroxylase deficiency
-
Current use of diuretics in hypertension
-
Cushing syndrome
-
Exogenous mineralocorticoids or glucocorticoids
-
Liddle syndrome
-
Renovascular hypertension
Causes of chloride-resistant alkalosis (urine chloride >20 mEq/L) without hypertension include the following:
-
Bartter syndrome
-
Gitelman syndrome
-
Severe potassium depletion
-
Current use of thiazides and loop diuretics
-
Hypomagnesemia
Other causes include the following:
-
Exogenous alkali administration - Sodium bicarbonate therapy in the presence of renal failure, metabolism of lactic acid or ketoacids
-
Milk-alkali syndrome
-
Hypercalcemia
-
Intravenous penicillin
-
Refeeding alkalosis
-
Massive blood transfusion
Epidemiology
Metabolic alkalosis is the most common acid-base disturbance observed in hospitalized patients, accounting for approximately 50% of all acid-base disorders.
Mortality rates have been reported as 45% in patients with an arterial blood pH of 7.55 and 80% when the pH was greater than 7.65. Prognosis also depends in part on the specific condition or circumstances that led to metabolic alkalosis.
-
Algorithm for metabolic alkalosis.
Tables

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Approach Considerations
- Serum Anion Gap
- Urine Sodium Ion Concentration
- Plasma Renin Activity and Aldosterone level
- Evaluations for Primary Hyperaldosteronism, Cushing Syndrome, and Apparent Mineralocorticoid Excess
- Evaluation for Congenital Adrenal Hyperplasia Variants
- Diuretic Screen, Adrenal Imaging, and Renovascular Hypertension Imaging
- Gene Analysis
- Show All
- Treatment
- Medication
- Questions & Answers
- References


