Practice Essentials
Burkitt lymphoma, or small noncleaved cell lymphoma, is a highly aggressive B-cell non-Hodgkin lymphoma (NHL) characterized by the translocation and deregulation of the c-myc gene on chromosome 8. [1] The 2016 World Health Organization (WHO) classification of lymphoid neoplasms recognizes Burkitt-like lymphoma with 11q aberration as a new provisional entity that lacks MYC rearrangements but resembles Burkitt lymphoma morphologically, to a large extent phenotypically, and by gene expression profiling. [2] See the image below.
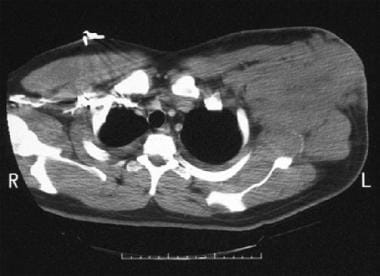 Computed tomography scan in a patient with a large, left-sided axillary mass from which a biopsy was obtained. Biopsy findings were consistent with small noncleaved cell non-Hodgkin lymphoma.
Computed tomography scan in a patient with a large, left-sided axillary mass from which a biopsy was obtained. Biopsy findings were consistent with small noncleaved cell non-Hodgkin lymphoma.
Signs and symptoms
Burkitt and Burkitt-like lymphomas have a rapid and aggressive clinical course with frequent bone marrow and central nervous system (CNS) involvement. These are considered to be medical emergencies and require immediate diagnostic and therapeutic intervention.
Endemic (African) Burkitt lymphoma (eBL) most commonly involves the jaw and facial bone (orbit) (> 50% of cases). Sporadic Burkitt lymphoma (sBL) most often presents as abdominal tumors with bone marrow involvement. Immunodeficiency-related Burkitt lymphoma cases usually as with nodal involvement with frequent bone marrow involvement.
Common findings and symptoms in patients with Burkitt lymphoma are summarized below.
Abdominal masses, which can cause abdominal pain and distention andascites
-
Nausea and vomiting
-
Loss of appetite, change in bowel habits, or both
-
Gastrointestinal bleeding
-
Signs and symptoms of acute abdomen
-
Intestinal perforation
-
Renal failure as a result of retroperitoneal disease and renal involvement or secondary to tumor lysis syndrome
Mandibular or maxillary mass
-
Most common presentation in eBL (maxillary > mandibular)
-
May involve the orbit; jaw involvement occurs much less frequently (15-20%)
CNS involvement
-
Meningeal infiltration, with or without cranial nerve (CN) involvement (frequently, CN III and CN VII); most common mode of presentation with CNS disease
-
Headaches, visual impairment, and paraplegia from spinal involvement; these may be the initial presenting features in some cases
"B" systemic symptoms
-
Uncommon but may be associated with other presenting symptoms (eg, fever, weight loss, night sweats, fatigue)
Other
-
Bone marrow involvemen
-
Painless lymphadenopathy (adults > children)
See Presentation for more detail.
Diagnosis
The least invasive procedure used to establish the diagnosis usually involves pathologic evaluation of the involved tissue biopsy. Patients with more than 25% bone marrow involvement are usually referred to as having Burkitt leukemia. Expedite the staging workup.
A diagnosis can sometimes be made by bone marrow aspiration and biopsy if the marrow is involved. If the marrow is not involved, diagnosis will require sampling lymph nodes or the involved extranodal site.
Staging
Because most patients will present with nodal or extranodal masses, different staging systems have been proposed.
The National Cancer Institute staging system is as follows:
-
A - Single solitary extra-abdominal site
-
AR - Intra-abdominal, more than 90% of tumor resected
-
B - Multiple extra-abdominal tumors
-
C - Intra-abdominal tumor
-
D - Intra-abdominal plus 1 or more extra-abdominal sites
The Ann Arbor system and St. Jude/Murphy staging (commonly used) consist of 4 stages.
Stage I is as follows:
-
Single tumor (extranodal)
-
Single anatomic area (nodal)
Stage II is as follows:
-
Single tumor (extranodal) with regional node involvement
-
Primary gastrointestinal tumor
-
Lymphoma involving nodal areas on the same side of the diaphragm
Stage IIR consists of completely resected intra-abdominal disease.
Stage III is as follows:
-
Lymphoma involving sites on opposite sides of the diaphragm
-
All primary intrathoracic tumors
-
All paraspinal or epidural tumors
-
Extensive intra-abdominal disease
Stage IV consists of any of the above, with CNS or bone marrow involvement (< 25%) at presentation.
The risk-adapted approach is used to treat most patients in the present-day clinical setting. Patients are divided into 2 broad groups, low- and high-risk patients. Low-risk patients have nonbulky disease (< 10 cm), early stage (I or II) disease, good performance status, and a normal lactate dehydrogenase (LDH) level. High-risk patients include all other patients.
Laboratory testing
-
Complete blood count with differentials and platelet count
-
Coagulation studies
-
Serum levels of electrolytes, uric acid, LDH, creatinine, beta2 microglobulin
-
Liver function tests
-
Tests for human immunodeficiency virus and hepatitis B
-
Histopathologic examination
-
Cytogenetic studies
-
Flow cytometry
Imaging studies
-
Head or spinal computed tomography (CT) scanning or magnetic resonance imaging: When neurologic signs/symptoms are present
-
Bone scanning and plain bone radiography: When bone symptoms are present
-
Chest, abdominal and pelvic CT scanning with intravenous (IV) contrast (omit contrast medium in the presence of renal insufficiency)
-
Echocardiography: For possible arrhythmias due to cardiac involvement
-
Multiple-gate acquisition scanning: For pre-chemotherapy evaluation of cardiac ejection fraction, particularly when anthracyclines will be used
Procedures
-
Unilateral bone marrow aspirate and biopsy for every patient with Burkitt lymphoma
-
Lumbar puncture as part of staging workup for cerebrospinal fluid involvement (defer this procedure in the presence of significant thrombocytopenia/coagulation defects)
-
Paracentesis or thoracentesis for cytogenetic studies of ascitic fluid or pleural effusion
See Workup for more detail.
Management
Chemotherapy is the mainstay of treatment for Burkitt lymphoma. Administer IV antibiotics for neutropenic fevers, and growth factors (granulocyte-macrophage colony-stimulating factor [GM-CSF] or granulocyte colony-stimulating factor [G-CSF]) to help decrease the duration of neutropenia. Surgery or radiation therapy has no role in the treatment of Burkitt lymphoma.
In general, 3 chemotherapy approaches are available for Burkitt lymphoma, as follows:
-
Intensive, short-duration regimens such as CODOX-M/IVAC (Magrath regimen) (cyclophosphamide, vincristine, doxorubicin, high-dose methotrexate / ifosfamide, etoposide, high-dose cytarabine) and the CALGB 9251 protocol
-
Long-duration chemotherapy similar to acute lymphoblastic leukemia treatment, such as hyper-CVAD (modified fractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone) and the Cancer and Leukemia Group B (CALGB) 8811 protocol
-
Combination regimens followed by autologous stem cell transplantation
-
Most current regimens add rituximab to previously established chemotherapy regimens
Other medications that may be used in patients with Burkitt lymphoma include the following:
-
Glucocorticoids (eg, prednisone)
-
Urate-oxidase enzymes (eg, rasburicase)
Supportive therapy
-
Prophylactic allopurinol and aggressive hydration with urine alkalinization: To reduce the risk of tumor lysis syndrome and uric acid nephropathy
-
Transfusions of leukodepleted and irradiated red blood cells or platelets (as clinically indicated): For anemia and thrombocytopenia
See Treatment and Medication for more detail.
Background
Historically, Burkitt lymphoma was termed malignant small noncleaved lymphoma in patients presenting with a solid tumor or nodal mass, whereas patients who had greater than 25% bone marrow involvement were considered to have French American British (FAB) L3 type acute lymphoblastic leukemia (L3 ALL). The 2016 World Health Organization (WHO) classification of lymphoid neoplasms identifies Burkitt lymphoma, in which TCF3 or ID3 mutations are present in up to about 70% of cases, and Burkitt-like lymphoma with 11q aberration, a new provisional entity that closely resembles Burkitt lymphoma but lacks MYC rearrangement and has some other distinctive features. [2]
The dysregulation and mutation of the c-myc oncogene that characterizes Burkitt lymphoma often results from a translocation between chromosomes 8 and 14, t(8;14)(q24;q32). Other translocations are also reported to cause c-myc overexpression, including t(2;8)(p12;q24) and t(8;22)(q24;q11). However, according to the WHO classification of lymphoid neoplasms, diagnosis of Burkitt lymphoma can still be made even in the absence of c-myc rearrangement, if other clinical, morphologic, and immunophenotypic findings support that diagnosis. [2]
Three distinct forms of Burkitt lymphoma are identified: (1) endemic (African), (2) sporadic, and (3) immunodeficiency-associated subtypes. Although these forms differ in their clinical presentation and their epidemiology, they share the same aggressive clinical behavior and are histologically identical.
The sporadic variant (sBL) is present in North America and Western Europe, and the endemic variant (eBL) is observed in equatorial Africa. Immunodeficiency-associated Burkitt lymphoma occurs most commonly in patients with human immunodeficiency virus (HIV) infection, but it has also been reported in the posttransplantation setting [3] as well as in congenital immunodeficiency patients. [4] Immunodeficiency-associated Burkitt lymphoma accounts for about 30% of lymphomas in HIV patients. [5]
Burkitt lymphoma was originally described in children, but this disease is also observed in adult patients. Burkitt lymphoma is one of the fastest growing malignancies in humans, with a growth fraction close to 100% and a doubling time of around 25 hours. [6]
Burkitt and Burkitt-like lymphomas have a rapid and aggressive clinical course, commonly presenting in children and young adults, with frequent bone marrow and central nervous system (CNS) involvement. These are considered to be medical emergencies and require immediate diagnostic and therapeutic intervention.
Historical information
Burkitt lymphoma is named after Dr. Dennis Burkitt, an Irish surgeon who worked in Kampala, Uganda, and noted a large number of African children affected with rapidly growing jaw tumors in areas where falciparum malaria was endemic. Dr. Burkitt described those tumors initially as a form of sarcoma. [7] It was not until 3 years later that the tumors were histologically identified by Burkitt and O’Conor as malignant lymphomas. [8] A few years later, Dr. Burkitt met and provided tumor samples to the pathologist Anthony Epstein. Dr. Epstein and his colleagues identified Epstein-Barr virus (EBV) from the lymphoma samples, which established a role for EBV in the pathogenesis of African Burkitt lymphoma. [9, 10, 11]
See also the following:
Etiology and Pathophysiology
The exact cause and pathophysiologic mechanisms leading to the development of Burkitt lymphoma are not known. Several theories exist. Epstein-Barr virus (EBV) and malaria infection as well as C-myc oncogene activation are briefly discussed in this section.
EBV and malaria infections
EBV is a member of the herpesvirus family that has been strongly implicated in the endemic form of Burkitt lymphoma (eBL). Virtually all patients with eBL are EBV positive, whereas only about 20% of sporadic (sBL) cases are associated with EBV. EBV tends to cause a latent infection of B lymphocytes, some of which evade the T-cell-mediated immune response and enter the germinal center. This subsequently results in excessive B cell proliferation. [2]
Malaria infection also probably plays a role in the pathogenesis of eBL, as it can lead to inhibition of EBV-specific immune response. [2] Like EBV, Plasmodium falciparum malaria is thought to contribute to eBL by inducing the expression of activation-induced cytidine deaminase (AID), an enzyme involved in the IGH/c-myc translocation. [12]
The exact mechanism of EBV-mediated lymphomagenesis, however, is not well understood, but evidence exists for a significant interaction between viral and cellular microRNA (miRNA) interfering with normal gene expression and translation. [13] EBV can be detected in 25-40% of immunodeficiency-associated cases. [2]
EBNA-1 (EBV nuclear antigen-1) and EBV-encoded RNAs have been shown to possess modest anti-apoptotic properties. Furthermore, EBNA-3A and EBNA-3C can inhibit the expression of the anti-apoptotic protein BCL-2. [14]
Genetic features
The classic t(8;14)(q24;q32) reciprocal translocation (85% of cases) results in the transposition of the c-myc proto-oncogene on chromosome 8 with one of the immunoglobulin heavy chain genes on chromosome 14, which results in activation of the c-myc gene and is considered responsible for tumor proliferation. Translocation t(8;14) is the most common, present in 80% of Burkitt lymphoma cases. In all the other cases, c-myc has been translocated close to one of the immunoglobulin light chain genes on chromosome 2 (kappa light chain) [t(8;2)] or 22 (lambda light chain) [t(8;22)].
Overproduction of the c-myc product may change the lymphocytes into cancer cells, but other gene mutations may be responsible for the progression of Burkitt lymphoma. [6] Abnormalities in the p53 gene and in death-associated protein kinase (DAP-kinase) has been shown to contribute to decreased apoptosis and to the pathogenesis of the disease. [15, 16]
C-myc is a leucine zipper transcription factor that affects different pathways regulating cell cycle, growth, adhesion, differentiation, and apoptosis. [17, 18] It is overexpressed via its juxtaposition with immunoglobulin gene enhancers. Genes such as cyclin D2, TRAP1, and HLA-DRB1 are induced with c-myc overexpression, whereas others like p21 and platelet-derived growth factor receptor-alpha (PDGFR-alpha) are consistently repressed, possibly playing a role in the pathogenesis of Burkitt lymphoma. [19]
E2F1 is a member of the E2F family of transcription factors that is involved in regulation of cell growth. Interestingly, in recent years, E2F1 was found to be overexpressed in most sporadic cases of Burkitt lymphoma. Furthermore, reduction of E2F1 expression led to decreased growth capacity in sBL cells in vitro. [20]
Mutations in the ID3 gene have been found in 34-68% of Burkitt lymphomas. The role of these inactivating ID3 mutations may be to significantly amplify the actions of the c-myc–immunoglobulin translocation, increas cell cycle progression and cellular proliferation in Burkitt lymphoma cells, [21, 22] In a study that involved genetic sequencing of 59 Burkitt lymphoma tumors, ID3 was one of 70 genes that were recurrently mutated in Burkitt lymphomas, including ID3, GNA13, RET, PIK3R1 and the SWI/SNF genes ARID1A and SMARCA4. [21]
Epidemiology
According to the SEER (Surveillance, Epidemiology and End Results) database, the yearly incidence of Burkitt lymphoma/leukemia in the United States across all ages, between 2001 and 2006 was 1759 cases. Most of these cases occurred in patients between ages 20 and 64 years. This disease constituted 0.4% of all lymphoid tumors.
About 30-40% of human immunodeficiency virus (HIV)–related non-Hodgkin lymphoma (NHL) cases are Burkitt lymphoma, and the rate did not seem to decrease with the advent of highly active anti-retroviral therapy (HAART), as Burkitt lymphoma typically affects patients with high CD4 counts (> 200/mm3). [23] This actually suggests that decreased immunity is not a risk factor in this variant of Burkitt lymphoma. (See HIV-1 Associated Opportunistic Neoplasms - CNS Lymphoma.)
Globally, Burkitt lymphoma is endemic in certain regions of equatorial Africa and other tropical locations between latitudes 10° south and 10° north. Incidence in these areas of endemic disease is 100 per million children. Epstein-Barr virus (EBV) infection is found in nearly all cases. In endemic areas, there seems to be a correlation with the geographic distribution of endemic malaria. [24, 25] (See Etiology and Pathophysiology.)
Although no racial predilection is reported, there is a sex and age predilection. Males are affected 2-3 times more often than females (male-to-female ratio, 2-3:1). According to the SEER database, between 2001 and 2006, the incidence of Burkitt lymphoma in adults (> 20 y) was roughly 5 times higher than the incidence of the disease in children (< 20 y).
The endemic form of the disease (eBL) is most common in children aged 4-7 years. Outside Africa, adults with the sporadic form (sBL) are typically younger than 35 years, whereas patients with Burkitt-like lymphoma (BLL) have a median age of 55 years. [26]
Prognosis
Approximately 90% of pediatric patients and up to 50-60% of adults with Burkitt lymphoma/Burkitt-like lymphoma (BL/BLL) treated with current intensive chemotherapy regimens have long-term disease-free survival. Hvelange et al identified genetic differences between adult and pediatric Burkitt lymphomas, which suggests age-related heterogeneity in pathogenesis that may explain the poorer prognosis in adult patients. [27]
Children
The prognosis of Burkitt lymphoma (BL) in children correlates with the bulk of disease at the time of diagnosis, as outlined below. Before the advent of aggressive therapeutic programs, children with Burkitt lymphoma (BL) died rapidly. With appropriate management of the metabolic consequences of rapid cell turnover and with combination chemotherapy and central nervous system (CNS) prophylaxis, the survival rate has been improved significantly (≤ 60%).
-
Patients with limited (A, AR; stage I and II) disease have an excellent prognosis, with a survival rate greater than 90%
-
Patients with more extensive disease (stage III and IV), especially involvement of the bone marrow and CNS, have a worse prognosis, but long-term survival rates of 50-90% can be achieved with more aggressive chemotherapy regimens
-
Patients with relapse disease have a long-term survival rate of 20-50%
-
For those who experience relapse, as many as 25% of children may be able to achieve a long-term disease-free survival through high-dose therapy with autologous hematopoietic stem-cell transplantation (HSCT). [28] The addition of rituximab to this therapeutic regimen may further increase the response rate. [29]
Adults
Adults with Burkitt lymphoma (BL), particularly those with advanced stage disease, do more poorly than children with the disease. [6, 30, 31, 32, 33, 34, 35] In addition, bone marrow involvement with complex cytogenetics has been noted to carry a worse prognosis in a retrospective observation [36] ; such complex cytogenetics have been reported to be more common among elderly patients. [37]
A prognostic scoring system developed in 2013 helps quantify the potential for cure in newly diagnosed adult patients with Burkitt lymphoma and helps stratify participants in future clinical trials. [38] Points are assigned as follows:
-
Age 40-59 years or Black race/ethnicity: 1 point
-
Age 60-79 years or stage III/IV disease: 2 points
-
Age 80 years and older: 4 points
The 4 risk groups based on the scoring system are as follows [38] :
-
Low-risk (5-year relative survival [RS]: 71%): 0-1 points
-
Low-intermediate (5-year RS: 55%): 2 points
-
High-intermediate (5-year RS: 41%): 3 points
-
High-risk (5-year RS: 29%): 4 or more points
Complications
Complications from disease include the following:
-
The likelihood of complications increases with the extent of the disease
-
Because of the extremely fast growth rate of Burkitt tumor cells, massive acute destruction of the tumor cells during initial chemotherapy may result in tumor lysis syndrome requiring renal dialysis (See Tumor Lysis Syndrome and Pediatric Tumor Lysis Syndrome.)
-
In the abdominal form of the disease, rapid tumor growth may result in obstruction; renal failure may occur from tumor infiltration of the kidneys
-
External compression of the ureters may cause obstructive uropathy; compression of loops of bowel may cause bowel obstruction
Complications from chemotherapy include the following:
-
Tumor lysis syndrome
-
Cardiomyopathy (anthracycline related)
-
Infections (during neutropenia post chemotherapy)
-
Gonadal dysfunction (chemotherapy-related sterility)
-
Secondary leukemias (exposure to alkylating agents), a late potential complication generally occurring more than 5 years after alkylator therapy
Patient Education
Provide information and support about the disease and the adverse effects of the drugs used to treat Burkitt lymphoma (BL) to patients and the family members, such as the following:
-
Secondary leukemias and myelodysplastic syndrome
-
Infertility
-
Possible anaphylactic reactions
-
Serious and potentially fatal infections
Inform patients who require high-dose chemotherapy and stem-cell transplantation that long-term complications of higher doses of chemoradiotherapy and a mortality rate of 3-5% from the conditioning regimen are possible
Young patients should be referred for counseling regarding fertility preservation, before chemotherapy, if possible.
Clearly explain transfusions (both red blood cells and platelets) and their associated complications.
Emotional support is very helpful to patients with cancer. Educating the medical personnel directly involved in patient care and the family members about emotional support for the patient is very important.
Patients should also be educated about the following:
-
Febrile neutropenia
-
Postchemotherapy thrombocytopenia and the tendency to bleed with minimal trauma
-
Chemotherapy-associated alopecia
-
Avoidance of pregnancy in women of childbearing age
-
Chemotherapy-induced nausea and vomiting
-
Chemotherapy-associated menstrual dysregulation (females) and the possibility of sexual dysfunction
-
Fatigue
-
Sperm banking
For patient education information, see Lymphoma and Burkitt Lymphoma.
-
Computed tomography scan in a patient with a large, left-sided axillary mass from which a biopsy was obtained. Biopsy findings were consistent with small noncleaved cell non-Hodgkin lymphoma.
-
Postchemotherapy computed tomography scan in a patient diagnosed with small noncleaved cell lymphoma (SNCCL) (same patient as in previous image). This image shows regression of a left axillary mass.
-
Coronal magnetic resonance imaging (MRI) section in a patient with large neck mass (same patient as in previous image). Biopsy findings showed Burkitt-like non-Hodgkin lymphoma (NHL). MRI was performed to assess for cord involvement.
-
Sagittal magnetic resonance imaging (MRI) section of the neck area showing a large mass invading the cervical spine with epidural encroachment (same patient as in the previous image). MRI was performed to rule out cord compression. The first image shows the gallium scan of this patient that correlates with the site of the tumor.
-
Right-sided pleural effusion in a patient with small noncleaved cell lymphoma (SNCCL) non-Hodgkin lymphoma.
-
Hematoxylin and eosin (H&E) stain. Sheets of monotonous-appearing lymphoid cells with one or more prominent nucleoli and an area of pale staining resulting from the presence of benign macrophages reveal a starry sky pattern.
-
The 2-dimensional flow cytometry demonstrates the highlighted cells to be CD5 negative and CD23 negative as well as lambda negative. Small noncleaved cell lymphoma (SNCCL) cells are typically CD19+, CD20+, CD22+, and CD10+.
Tables

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Approach Considerations
- Chemotherapy Overview
- CODOX-M/IVAC Regimen (Magrath Regimen)
- CALGB 9251 Regimen
- Hyper-CVAD Regimen
- Rituximab
- Evaluation of Treatment Response
- Relapsed/Refractory Burkitt Lymphoma
- Stem Cell Transplantation
- Surgical Intervention
- Long-Term Monitoring
- Special Considerations
- Show All
- Guidelines
- Medication
- Questions & Answers
- Media Gallery
- References








