Practice Essentials
Luteinizing hormone (LH) is a peptide produced by the anterior pituitary, along with follicle-stimulating hormone (FSH), in response to gonadotropin-releasing hormone (GnRH) from the hypothalamus. LH and FSH play central roles in the hypothalamic-pituitary-gonadal axis, and, thus, conditions related to LH and FSH deficiency can be caused by pathology of either the hypothalamus or the pituitary (see the image below). Idiopathic hypogonadotropic hypogonadism (IHH) is a rare condition that results from abnormal production of GnRH, leading to LH and FSH deficiency. IHH associated with anosmia is known as Kallmann syndrome. LH deficiency can also occur due to stress-related hypogonadotropic hypogonadism, LH subunit mutations, congenital pituitary hormone deficiency (CPHD), pituitary tumors, inflammation, vascular accidents, and pregnancy-related hemorrhagic shock (Sheehan syndrome). LH deficiency can manifest as delayed puberty in males or females, primary or secondary amenorrhea in females, or reproductive tract abnormalities. Careful analysis of a patient’s presenting symptoms, reproductive history, future desire for fertility, and hormonal profile are necessary to determine the cause of LH deficiency and, thus, the most appropriate treatment.
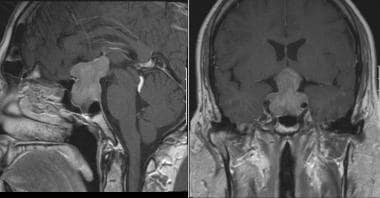 MRI of pituitary macroadenoma.
MRI of pituitary macroadenoma.
Signs and symptoms of LH deficiency
Patients with Kallmann syndrome and those with idiopathic hypogonadotropic hypogonadism lack secondary sex characteristics. Patients with Kallmann syndrome are also affected by either anosmia or severe hyposmia.
Stress-related hypogonadotropic hypogonadism presents in women as amenorrhea.
Pituitary dysfunction in women can result in irregular menses or amenorrhea. In the presence of hyperprolactinemia, approximately one third of women have galactorrhea as well. Men with hyperprolactinemia can present with hypogonadism, impotence, infertility, and/or galactorrhea.
See Presentation for more detail.
Diagnosis of LH deficiency
Laboratory studies
The basic laboratory evaluation for females or males suspected of having LH deficiency includes serum levels of thyroid-stimulating hormone (TSH), prolactin (PRL), LH, FSH, and estradiol. Low or normal LH and FSH levels in the presence of low estradiol suggest a hypothalamic disorder. A pituitary disorder is most commonly associated with elevated PRL levels.
Imaging studies
When hypothalamic or pituitary dysfunction is suspected, the most important imaging study is magnetic resonance imaging (MRI) of the head to detect a tumor or other abnormality.
See Workup for more detail.
Management of LH deficiency
Treatment of hypogonadotropic hypogonadism depends on the gender and age of the patient as well as their desire for current fertility.
Men and women with LH deficiency secondary to pituitary dysfunction require treatment depending on the presenting symptoms and associated hormonal disorders.
See Treatment and Medication for more detail.
Background
Structure and genetics
GnRH is a decapeptide secreted by neurons in the arcuate nucleus of the hypothalamus and released into the pituitary portal circulation. LH and FSH are produced by gonadotrope cells located in the anterior pituitary gland. The gonadotrope cells release LH and FSH in a pulsatile fashion with frequency and amplitude varying during the menstrual cycle, approximately every hour when stimulated by GnRH. Once released into the systemic circulation, both LH and FSH stimulate the gonads of females and males to release steroid hormones. [1]
LH is a glycoprotein dimer composed of 2 glycosylated noncovalently-linked subunits designated alpha and beta. The alpha subunit is composed of 92 amino acids and is encoded on the long arm of chromosome 6. The beta subunit is 121 amino acids and is encoded on the long arm of chromosome 19.
The alpha subunit of LH is biologically identical to 3 other hormones: FSH, thyroid-stimulating hormone (TSH), and human chorionic gonadotropin (hCG). The unique beta subunit determines LH immunologic and biologic activity. The half-life of LH and FSH are 30-60 minutes and 3-5 hours, respectively. LH and hCG act via the luteinizing hormone chorionic gonadotropin receptor (LHCGR). This transmembrane G protein-coupled receptor acts with ligand binding to enhance binding of guanosine 5'-triphosphate (GTP) to the G protein with subsequent disassociation from the receptor followed by activation of the cyclic adenosine monophosphate (cAMP) system. Mutations of the LH receptor can lead to inactivity or constitutive activity of LH. [2] Complete inactivating mutations in the genetic male lead to female external genitalia and lack of pubertal development, while partial inactivating mutations may result in micropenis or hypospadias. Genetic females with LHCGR mutations have normal internal and external genitalia, normal pubertal development but amenorrhea or irregular cycles, infertility, and ovarian cysts secondary to anovulation. [3] Activating LHCGR mutations results in male precocious puberty and Leydig cell hyperplasia. Interestingly, females with activating mutations have no symptoms, even though increased thecal androgen production would be expected. [4]
Menstrual cycle
In the female, coordinated LH and FSH stimulate the ovary to secrete estradiol, progesterone, and androgens in a cyclic manner. During the follicular phase, LH primarily stimulates theca cells to produce androgens. These androgens are aromatized to estradiol in the granulosa cells of the maturing ovarian follicle under the influence of FSH. At mid-cycle, estradiol has a positive feed-back effect on the hypothalamus, which triggers a dramatic spike in the release of LH. This LH surge initiates ovulation, resumption of oocyte meiosis, and the conversion of the mature follicle into the corpus luteum, which then produces progesterone primarily under the influence of LH.
During the luteal phase, LH continues to stimulate the corpus luteum to produce estradiol and progesterone. These steroid hormones act upon the endometrium to make it receptive to embryo implantation. If pregnancy occurs, placental trophoblasts secrete hCG, which stimulates the corpus luteum to continue production of estrogen and progesterone in support of the pregnancy. In the absence of pregnancy, decreasing LH levels cause corpus luteum regression approximately 2 weeks after ovulation. The consequential drop in progesterone results in menstruation. [5]
Spermatogenesis
In the male, both LH and FSH are required for spermatogenesis. LH stimulates Leydig cells to convert cholesterol to testosterone. Testosterone and FSH, in turn, modulate Sertoli cells, which serve as "nurse" cells for spermatogenesis within the lumen of the seminiferous tubules. Clinically, only FSH is mainly used as a marker of testicular dysfunction. [6]
Causes
LH deficiency may be due to genetic mutations or secondary to hypothalamic/pituitary acquired abnormalities from tumors, infiltrating diseases, or radiation therapy. During embryonic development, GnRH neurons originate in the nasal placode and share an embryonic origin with olfactory neurons. Multiple gene mutations have been identified with varying pathology of gonadotropin deficiency with olfactory defects, midline facial defects, and renal abnormalities.
Hypothalamic causes of LH deficiency
The nomenclature for hypothalamic causes of gonadotropin deficiency can be confusing with the abbreviation of IHH standing for both idiopathic hypogonadotropic hypogonadism (also called congenital hypogonadotropic hypogonadism [CHH]) and for isolated hypogonadotropic hypogonadism (also called congenital gonadotropin-releasing hormone deficiency [IGD]). This may be due to most cases of hypogonadotropic hypogonadism remaining unexplained prior to molecular diagnoses. Even now, a molecular diagnosis is not typically sought in the absence of other symptoms.
IHH and Kallman syndrome
-
Epidemiology: IHH represents a class of genetic disorders related to abnormalities in production or activity of a hypothalamic peptide hormone, gonadotropin releasing hormone (GnRH), that is essential to human reproduction. Hypogonadotropic hypogonadism has an overall incidence of approximately 1:10,000 to 1:86,000 men and women. There are two classes of IHH: (1) IHH associated with normal olfaction (normosomic IHH); or (2) IHH associated with anosmia, which is commonly termed Kallman syndrome and was first described by Franz Hosef Kallman in 1944. [7] Approximately 66% of the cases of IHH are associated with Kallman syndrome. Kallman syndrome occurs sporadically in 60% of patients, but can be genetically transmitted as an X-linked, autosomal dominant or autosomal recessive condition depending on the specific causative gene mutation. [8] Over 35 gene abnormalities have been associated with IHH, and the list continues to increase. [9]
-
Pathophysiology: Kallmann syndrome results from the congenital absence of GnRH-producing neurons in the hypothalamus. During embryogenesis, olfactory axonal and GnRH neurons from the olfactory placode fail to migrate to the hypothalamus. In the absence of GnRH, the pituitary gonadotrope cells are not signaled to produce LH and FSH, ultimately leading to lack of sex hormone production by the gonads. [8] In males, the most common cause of Kallmann syndrome is a X-linked mutation of the KAL1 gene resulting in deficient or abnormal cell adhesion protein Anosmin-1. Anosmin-1 participates in olfactory bulb localization and migration of GnRH neurons from the forebrain to the hypothalamus. In the absence of GnRH, the pituitary gonadotrope cells are not signaled to produce LH and FSH, ultimately leading to lack of sex hormone production by the gonads. [8] Given tissue-specific expression of KAL-1 in corticospinal tract, mesonephric tubules, and ureteric bud, there is an increased prevalence of abnormalities of the neurologic system, facial abnormalities, and urinary tract (eg, renal agenesis). In females, common causes of Kallmann include mutations in fibroblast growth factor receptor 1 and fibroblast growth factor 8.
-
Symptoms: There is a broad spectrum of clinical presentation of IHH. Presenting symptoms can range from partial completion of puberty to complete absence of sexual development. Both females and males with Kallmann syndrome usually present with anosmia and delayed puberty. Females present with primary amenorrhea, and some males present with micropenis.
-
Diagnosis: Essential to the diagnosis of Kallman syndrome is the lack of endogenous LH during blood sampling. [7] Laboratory evaluation reveals low LH and FSH levels and normal karyotypes.
-
Treatment: Hormone replacement therapy (estrogen for females and testosterone for males) is used to induce sexual maturation and minimize the long-term risk of osteoporosis. In females, after completion of breast development, progesterone or a synthetic progestin is added to protect the endometrium from hyperplasia secondary to long-term unopposed estrogen exposure. When fertility is desired, the treatment consists of either GnRH, given by a subcutaneous pump (not available in the United States) or exogenous gonadotropins given by injection. Women with Kallmann syndrome do not ovulate when given clomiphene citrate, which relies on an intact hypothalamic-pituitary-gonadal axis. Likewise, maintenance therapy with clomiphene citrate does not appear to increase testosterone secretion or sperm production in men with Kallmann syndrome.
Stress-related hypogonadotropic hypogonadism
-
Epidemiology: Stress-related hypogonadotropic hypogonadism, also frequently referred to as hypothalamic or functional amenorrhea, accounts for more than 30% of secondary amenorrhea in reproductive-aged women. [8] Among childhood cancer survivors who were treated with hypothalamic-pituitary radiotherapy, the estimated prevalence of LH/FSH deficiency is 10.6%. [10]
-
Pathophysiology: Hypothalamic suppression can occur in women under physical or metabolic stress. Stress-related hypothalamic suppression is most commonly related to prolonged strenuous physical exercise and extreme weight loss, particularly in the context of eating disorders, such as anorexia nervosa and bulimia. [11] These conditions cause an elevation of corticotropin-releasing hormone (CRH), which leads to increased central beta-endorphin release, inhibiting pulsatile GnRH release from the hypothalamus. [12] Alterations of the normal hypothalamic-pituitary-ovarian axis can include a lower LH pulse amplitude and a higher pulse frequency than in eumenorrheic women. [12]
-
Symptoms: Suppression of GnRH release in women results in decreased secretion of LH and FSH (ie, hypogonadotropic hypogonadism), manifesting as amenorrhea and hypoestrogenemia. [13] Ongoing hypothalamic suppression can lead to serious consequences such as osteoporosis and bone fractures in these women.
-
Diagnosis: Initial testing for secondary amenorrhea includes serum levels of thyroid-stimulating hormone (TSH), prolactin (PRL), FSH, LH, and estradiol. Low or normal LH and FSH levels in the presence of low estradiol suggest a hypothalamic disorder with stress-related hypogonadotropic hypogonadism the most likely cause after other diagnoses are excluded.
-
Treatment: Treatment of the primary cause is recommended, as most causes are reversible. Estrogen and progesterone/progestin therapy is recommended to prevent excessive bone loss. Contraception is recommended, as this can be a fluctuating condition with episodic unpredictable ovulation. If fertility is desired, patients are encouraged to maintain good nutrition and a normal body weight, but may also require ovulation induction with exogenous gonadotropins or pulsatile GnRH therapy. [12]
Pituitary causes of LH deficiency
The anterior pituitary produces a number of important peptide hormones, including LH, FSH, TSH, adrenocorticotropic hormone (ACTH), prolactin (PRL), and growth hormone (GH). LH deficiency can result from a myriad of anterior pituitary dysfunctions including congenital pituitary hormone deficiency (CPHD) caused by inherited or spontaneous gene mutations, pituitary tumors, inflammation, vascular accidents, and pregnancy-related hemorrhagic shock (Sheehan syndrome).
Congenital pituitary hormone deficiency (CPHD)
-
Epidemiology: The incidence of congenital pituitary hormone deficiency (CPHD) is 1 in 4000 to 10,000, with most cases occurring sporadically. [14] A genetic cause has been identified in some familial cases, but most cases do not have a specific genetic mutation identified.
-
Pathophysiology: Patients with CPHD lack one or more pituitary hormones due to congenital or spontaneous gene mutations. [15] Only about 16% of cases are associated with a mutation in a known gene, with PROP1 (Homeobox protein prophet of PIT-1) mutations occurring most frequently.
-
Symptoms: Symptoms may reflect the developmental relationship between the pituitary and nearby structures. The pituitary develops closely with the eye, optic nerve, ear, nose, and cranial nerve ganglia; thus, abnormalities of these structures as well as midline craniofacial defects (cleft lip or palate) can occur with CPHD depending on the specific mutations involved. Mutations in transcription factors involved in early pituitary development cause most syndromic forms of CPHD. [15] Males with LH/FSH deficiency may present with micropenis and undescended testes, whereas female genitalia are unaffected.
-
Diagnosis: Testing should include LH, FSH, TSH, and prolactin levels, ACTH stimulation testing, and GH levels (newborns) or IGF-1 levels, as well as brain MRI. Abnormal MRI findings can include a mildly or severely hypoplastic pituitary gland, interrupted or hypoplastic pituitary stalk, and/or an ectopic posterior pituitary. [15]
-
Treatment: Patients with CPHD need hormone replacement tailored to their specific hormone deficiencies. Pulsatile GnRH therapy may help normalize LH, FSH, and testosterone levels in some males (about 60%) with CPHD. [16]
Pituitary dysfunction
-
Epidemiology: Pituitary dysfunction is found in approximately one third of women with secondary amenorrhea. Of these, approximately one third have a pituitary tumor, and one third of those with a tumor have associated galactorrhea. Overall, the prevalence of clinically significant pituitary adenomas is less than 0.01% of the population. [8]
-
Pathophysiology: Pituitary gonadotropin suppression may be due to suppression of GnRH from neurohormones related to stress described above or to hyperprolactinemia or to destruction of pituitary cells. Hyperprolactinemia is a common hormonal abnormality associated with anterior pituitary dysfunction. Women with high levels of serum prolactin (>20-25 ng/mL) often can develop galactorrhea, and some develop amenorrhea and hypoestrogenism. The amenorrhea related to hyperprolactinemia is caused by alterations in the normal release and pulsatility of GnRH as well as subsequent alterations in LH/FSH secretion and the LH surge. [17] Causes of hyperprolactinemia include pituitary adenomas, primary hypothyroidism, hypothalamic dysfunction, chronic renal insufficiency, and chest wall trauma or herpes zoster infection. Many medications such as antipsychotics, estrogen, antihypertensives, metoclopramide, and cimetidine can also cause hyperprolactinemia. Other pituitary causes of LH deficiency that are often concurrent with other pituitary hormone deficiencies include empty sella syndrome, previous meningitis or encephalitis, hydrocephalus, infiltrating diseases such as tuberculosis, sarcoidosis, and hemochromatosis and intracranial tumors.
-
Symptoms: There are a myriad of symptoms depending on the etiology of the pituitary dysfunction. Regardless of cause, gonadotropin deficiency results in classic symptoms of irregular cycles or amenorrhea in women and sexual dysfunction/infertility in men. Consequential lack of estrogen is associated with bone loss and vaginal symptoms of dryness and irritation. In males, lack of testosterone leads to decreased body hair growth, decreased muscle mass, bone loss, and possibly gynecomastia. With hyperprolactinemia, approximately 33% of women will experience galactorrhea. Loss of other pituitary hormones can result in a multitude of symptoms, not covered in this review. Intracranial tumors may cause headaches depending on size and location, along with loss of balance, vision changes, mental changes, and seizures.
-
Diagnosis: When evaluating hyperprolactinemia, it is important to rule out hypothyroidism prior to proceeding with imaging. When hypothalamic or pituitary dysfunction is suspected, the most important imaging study is magnetic resonance imaging (MRI) of the head to detect a tumor or other abnormality.
-
Treatment: Treatment depends on the etiology and consequences of the disorder. Hyperprolactinemia secondary to hypothyroidism is corrected with thyroid replacement, whereas a causative medication may be changed. Increased prolactin secondary to a pituitary adenoma may be corrected with a dopamine agonist such as cabergoline or bromocriptine. In cases where the primary cause of the hypogonadotropic hypogonadism cannot be corrected, then the emphasis is on normalization of sex hormone levels with estrogen and progesterone/progestin in females and testosterone in males.
Differential diagnosis
Therefore, the causes of LH deficiency can be summarized as follows:
Kallmann syndrome
-
Genetic
Hypogonadotropic hypogonadism
-
Genetic
-
Idiopathic
-
Prolonged strenuous exercise
-
Anorexia nervosa/bulimia
-
Starvation
Pituitary dysfunction
-
Congenital pituitary hormone deficiency
-
Hyperprolactinemia
-
Intracranial tumors
-
Pituitary tumors
-
Pituitary infarction
-
Infiltrating diseases of the pituitary
Epidemiology
United States statistics
Hypogonadotropic hypogonadism has an overall incidence of approximately 1:10,000 to 1:86,000 men and women. Two thirds of the time, it is associated with anosmia (ie, Kallmann syndrome).
Stress-related hypogonadotropic hypogonadism accounts for more than 30% of secondary amenorrhea in reproductive-aged women. [8]
Pituitary dysfunction is found in approximately one third of women with secondary amenorrhea. Of these, approximately one third have a pituitary tumor, and one third of those with a tumor have associated galactorrhea. Overall, the prevalence of clinically significant pituitary adenomas is less than 0.01% of the population. [8]
Among childhood cancer survivors who were treated with hypothalamic-pituitary radiotherapy, the estimated prevalence of LH/FSH deficiency is 10.6%. [10]
International statistics
LH deficiency is not unique to any particular country or race.
Race-, sex-, and age-related demographics
LH deficiency occurs in all races. No racial predilection exists.
Kallmann syndrome is 7 times more common in males than in females. Hypogonadotropic hypogonadism occurs in both men and women, but adult onset is more common for women. Pituitary dysfunction occurs in both men and women.
Kallmann syndrome and genetic forms of IHH are usually diagnosed in children with delayed puberty. Adult onset IHH can occur at any age. Stress-related hypogonadotropic hypogonadism is most common in young women. Pituitary adenomas occur at all ages, but the incidence of diagnosis peaks at approximately 40 years of age.
Prognosis
Most causes of LH deficiency are irreversible. However, with appropriate hormone replacement therapy, fertility and a normal life expectancy can be anticipated.
Morbidity/mortality
The primary medical risks of LH deficiency are abnormal development, sexual dysfunction, and infertility. If untreated, resulting hypogonadism also puts patients at risk for medical conditions associated with low testosterone in males and low estrogen in females, including osteoporosis and bone fractures.
Complications
LH deficiency results in infertility and decreased sex hormones if untreated. Complications associated with the secondary lack of estrogen or testosterone can be avoided by replacement hormone therapy. Hypothalamic and pituitary anomalies can result in other hormonal deficiencies (eg, thyroid, adrenal) that can adversely affect health.
Patient Education
Patients need to be educated about the incidence, pathophysiology, and treatment of their specific condition.
-
MRI of pituitary macroadenoma.






